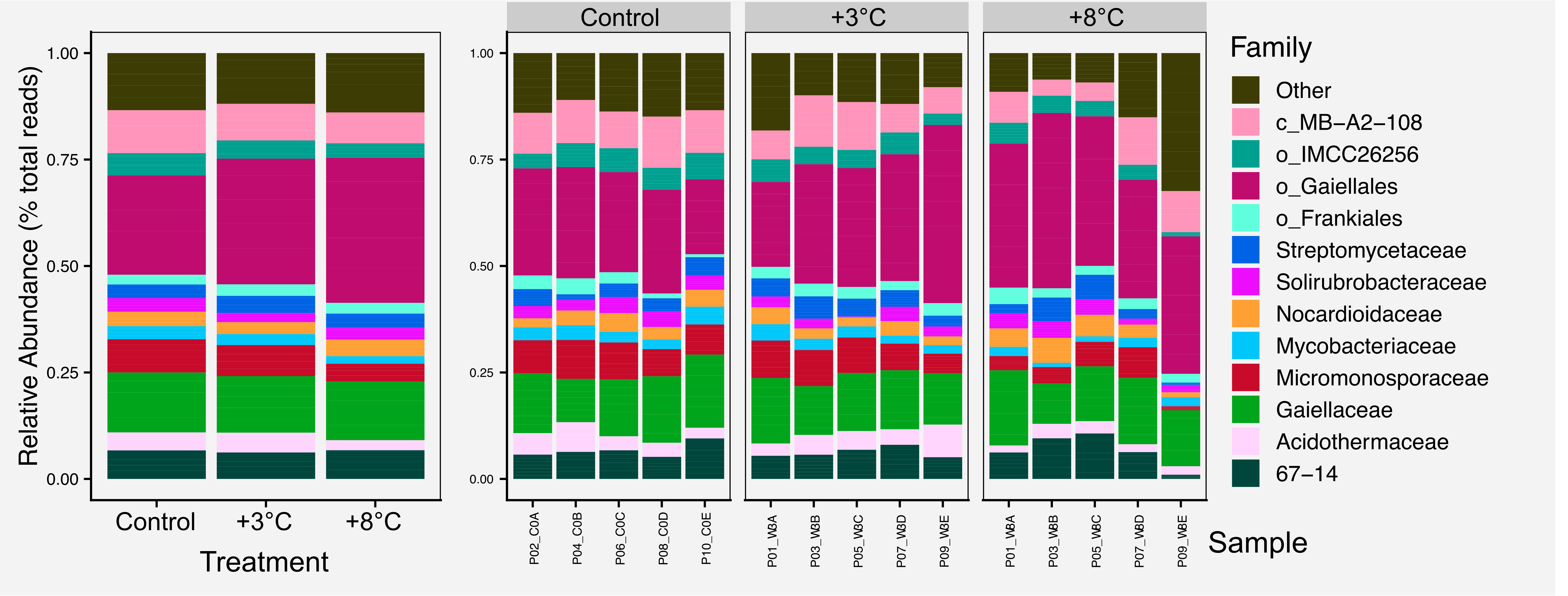
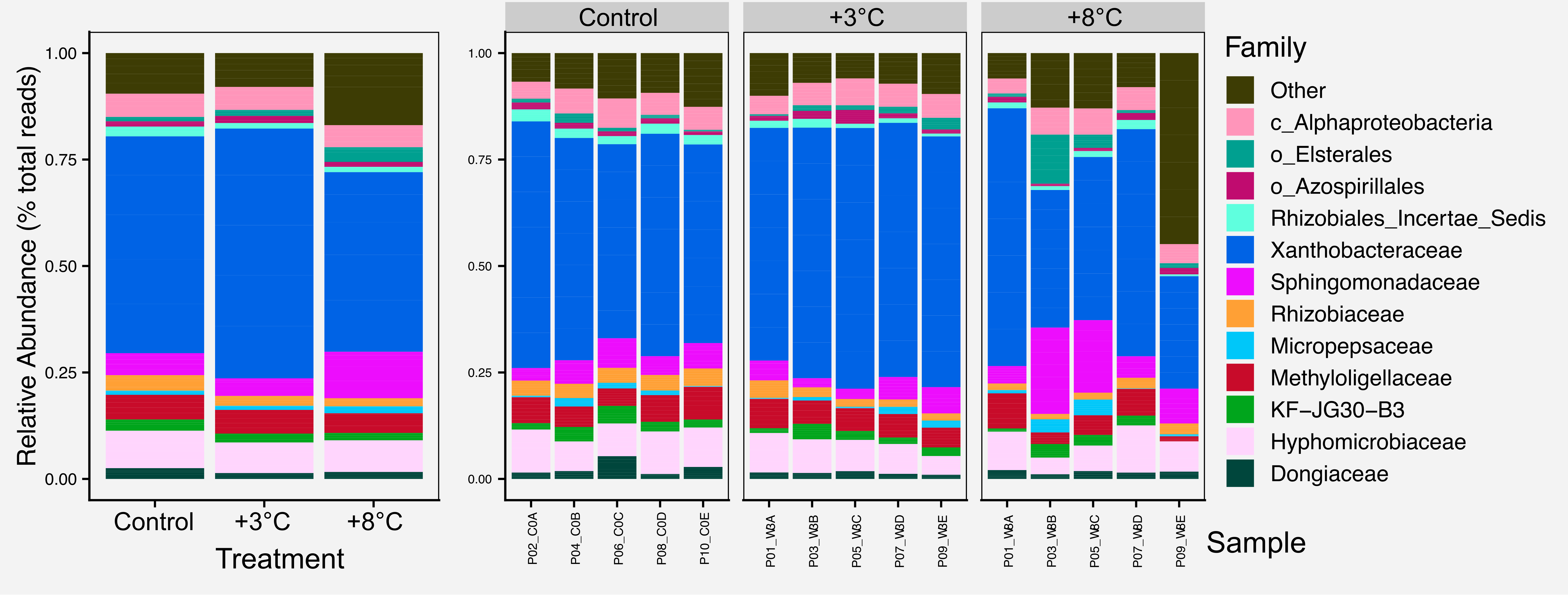
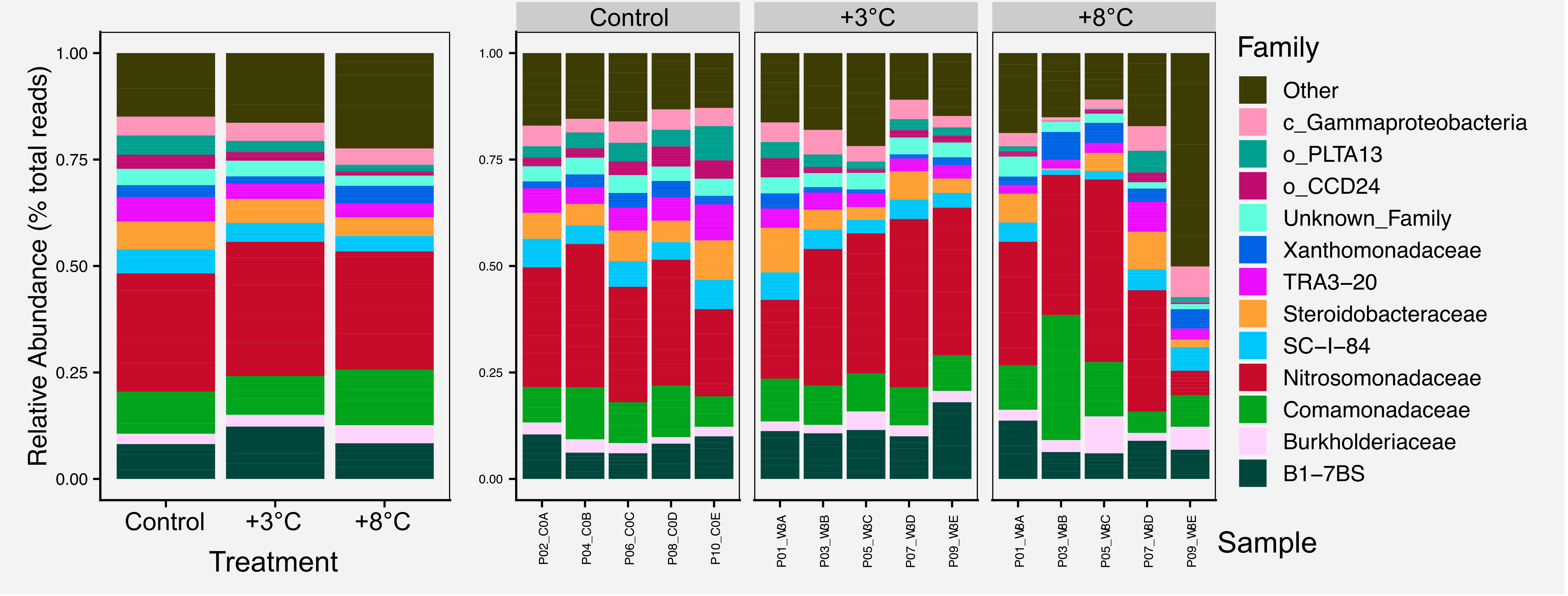
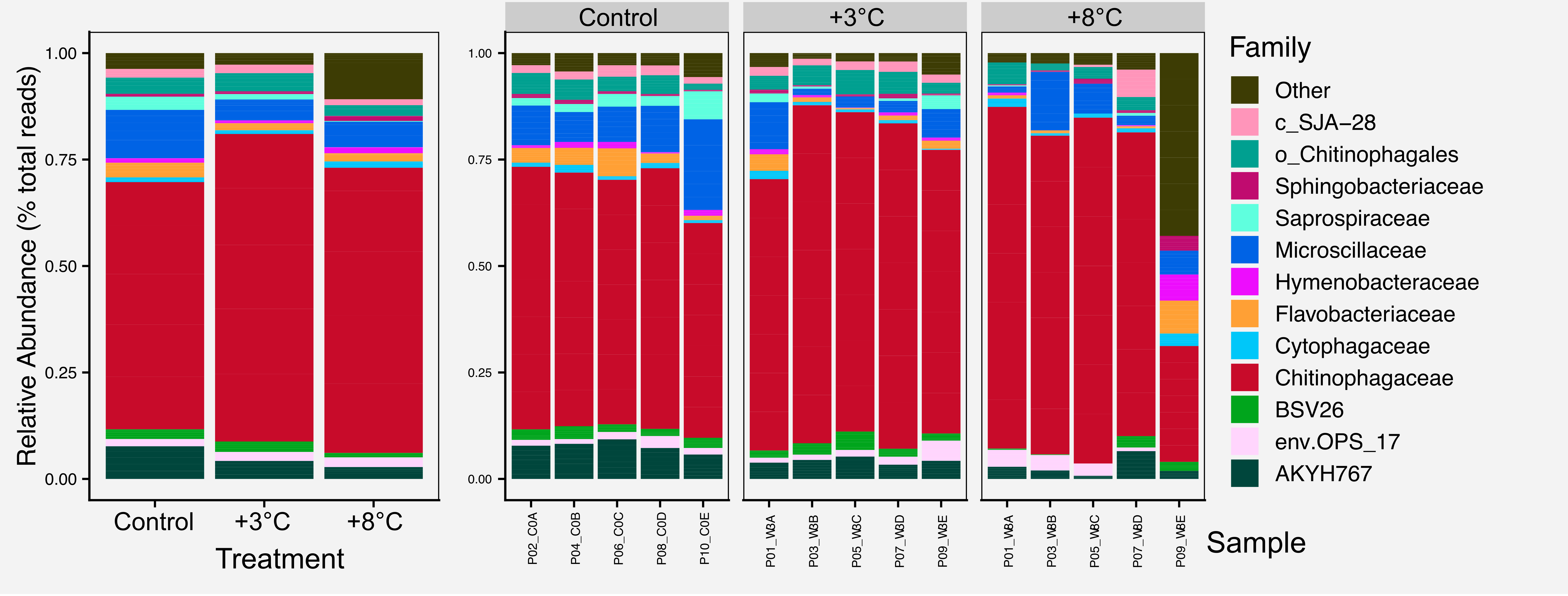
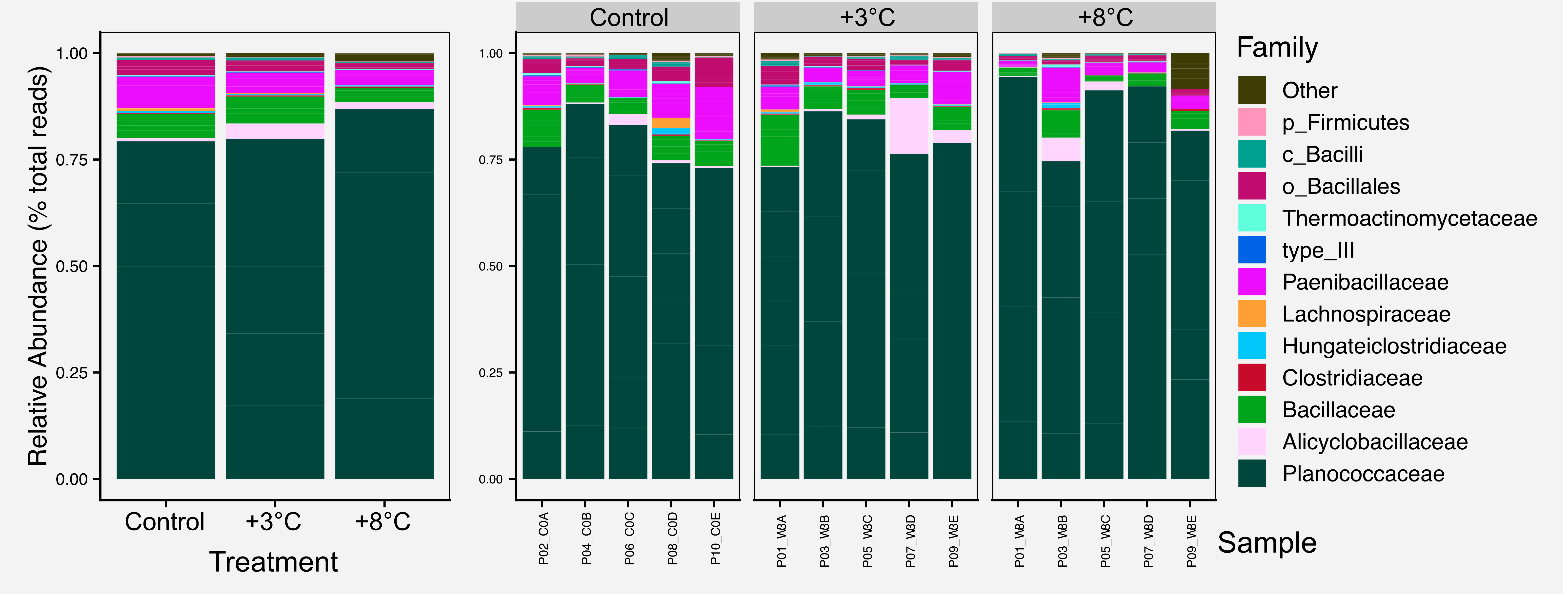
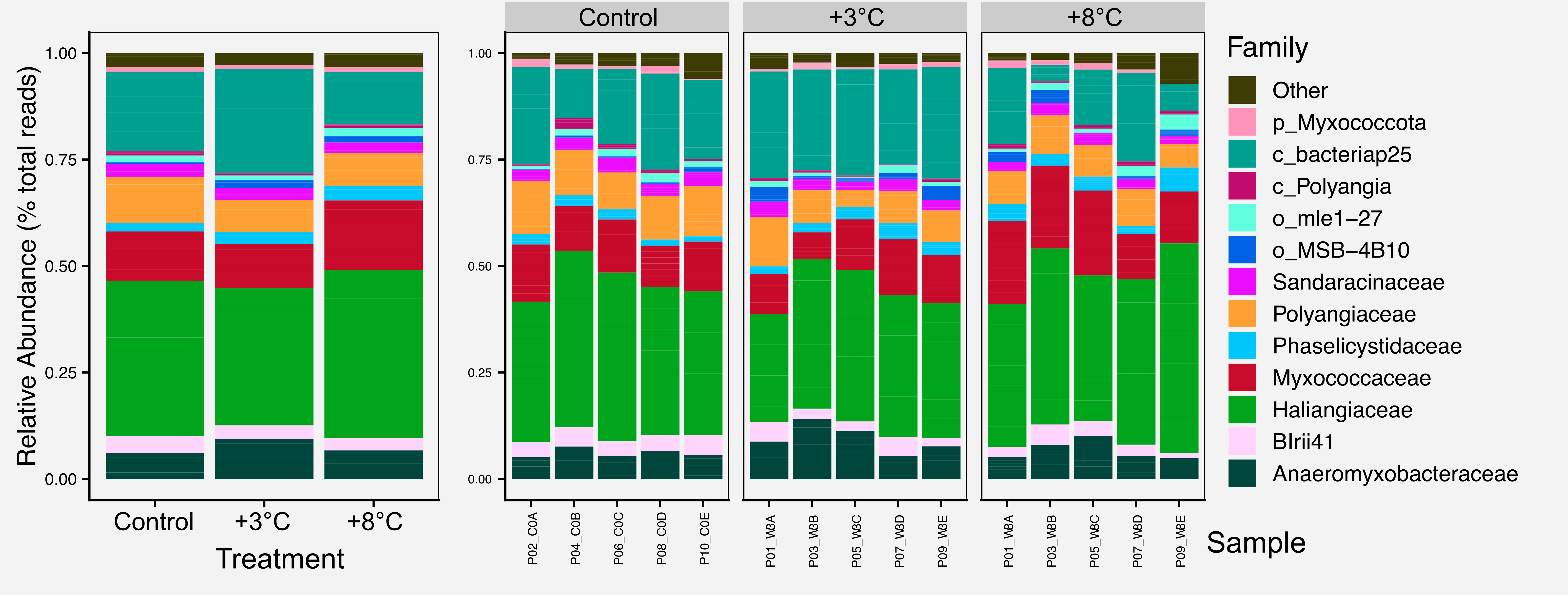
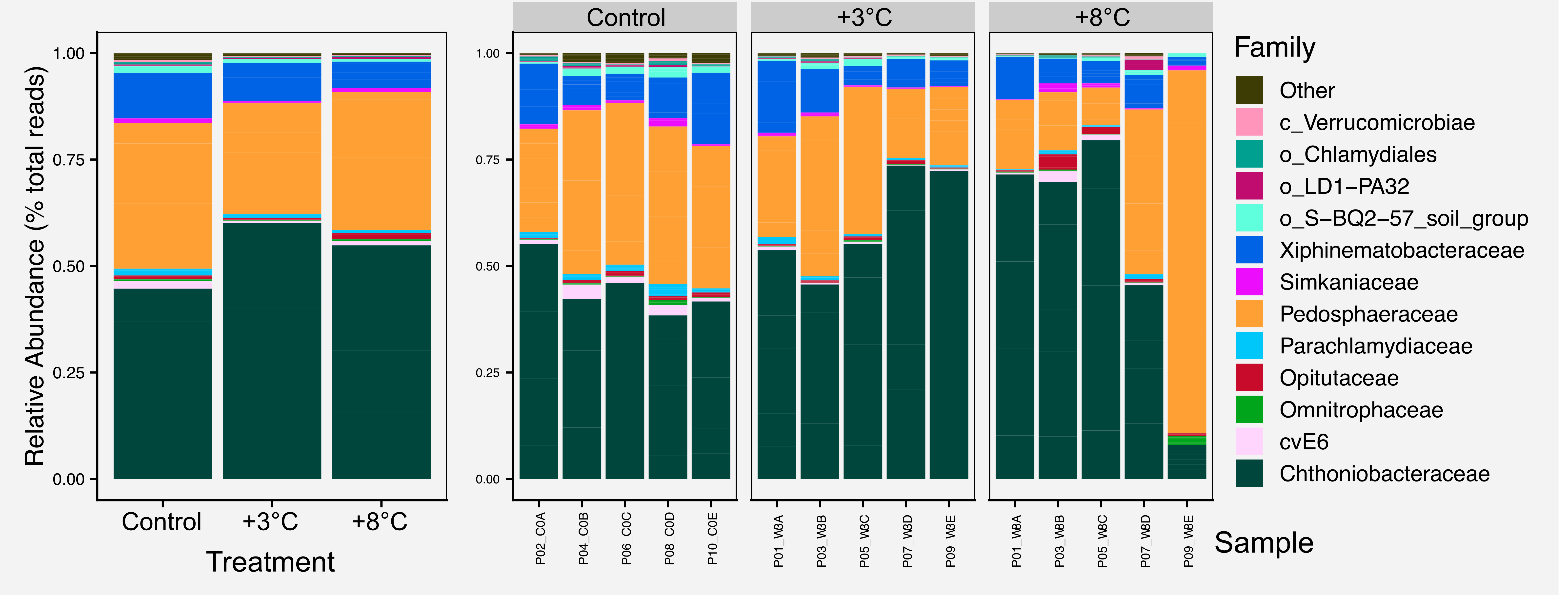
For most figures we include the raw figure generated in R, the post-processed figure, plus the code and data needed to generate the figure.
Main Paper
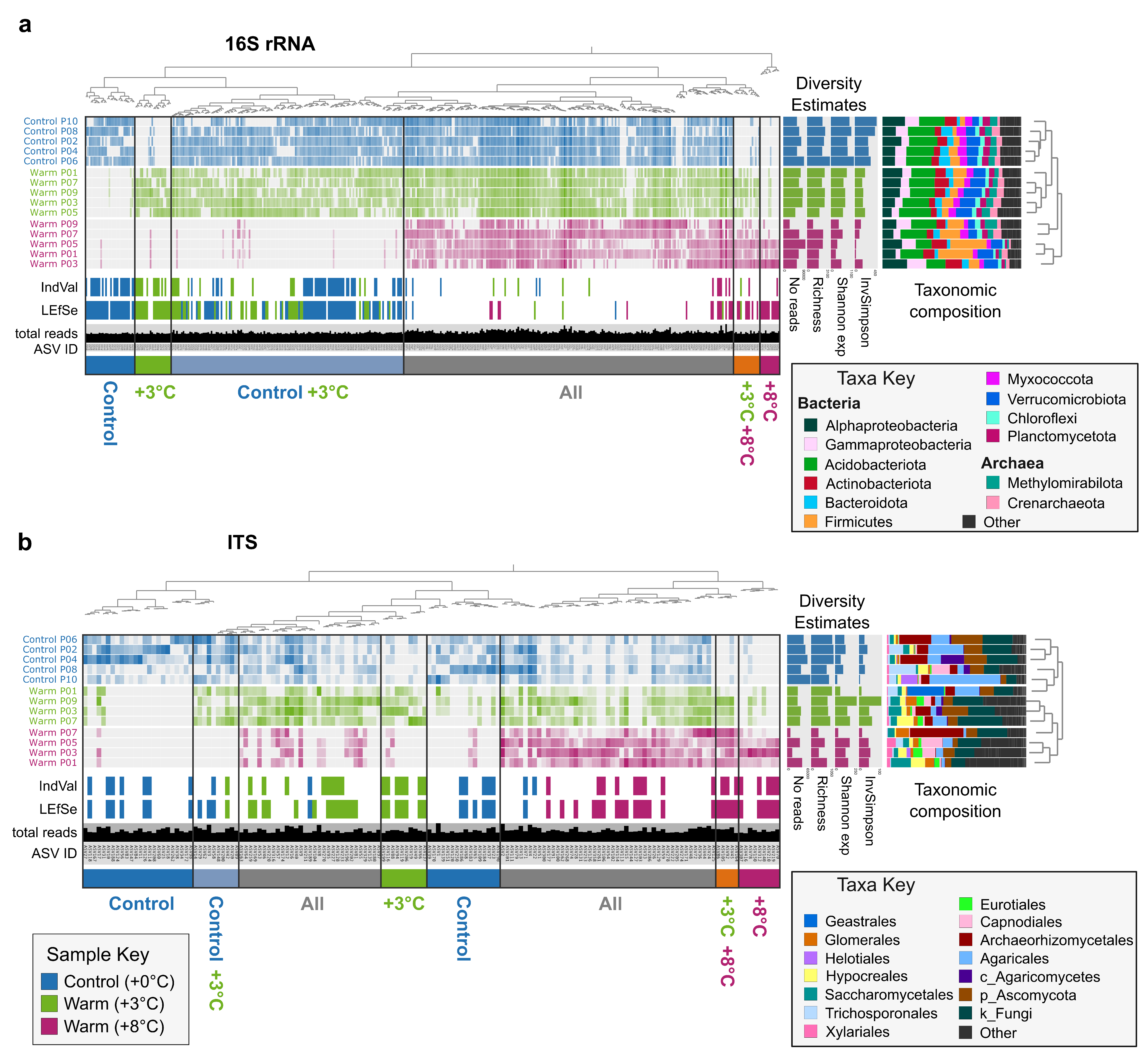
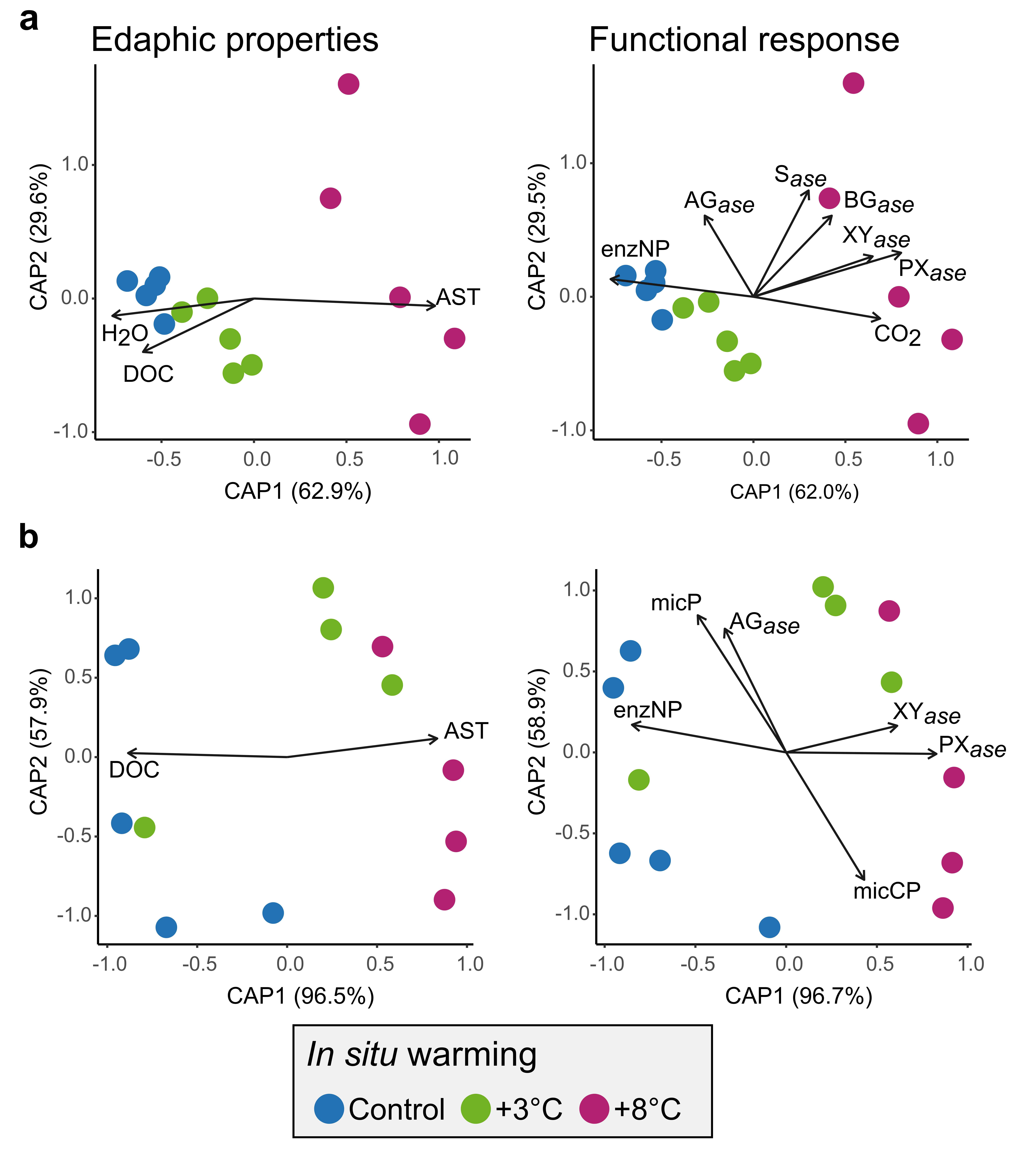
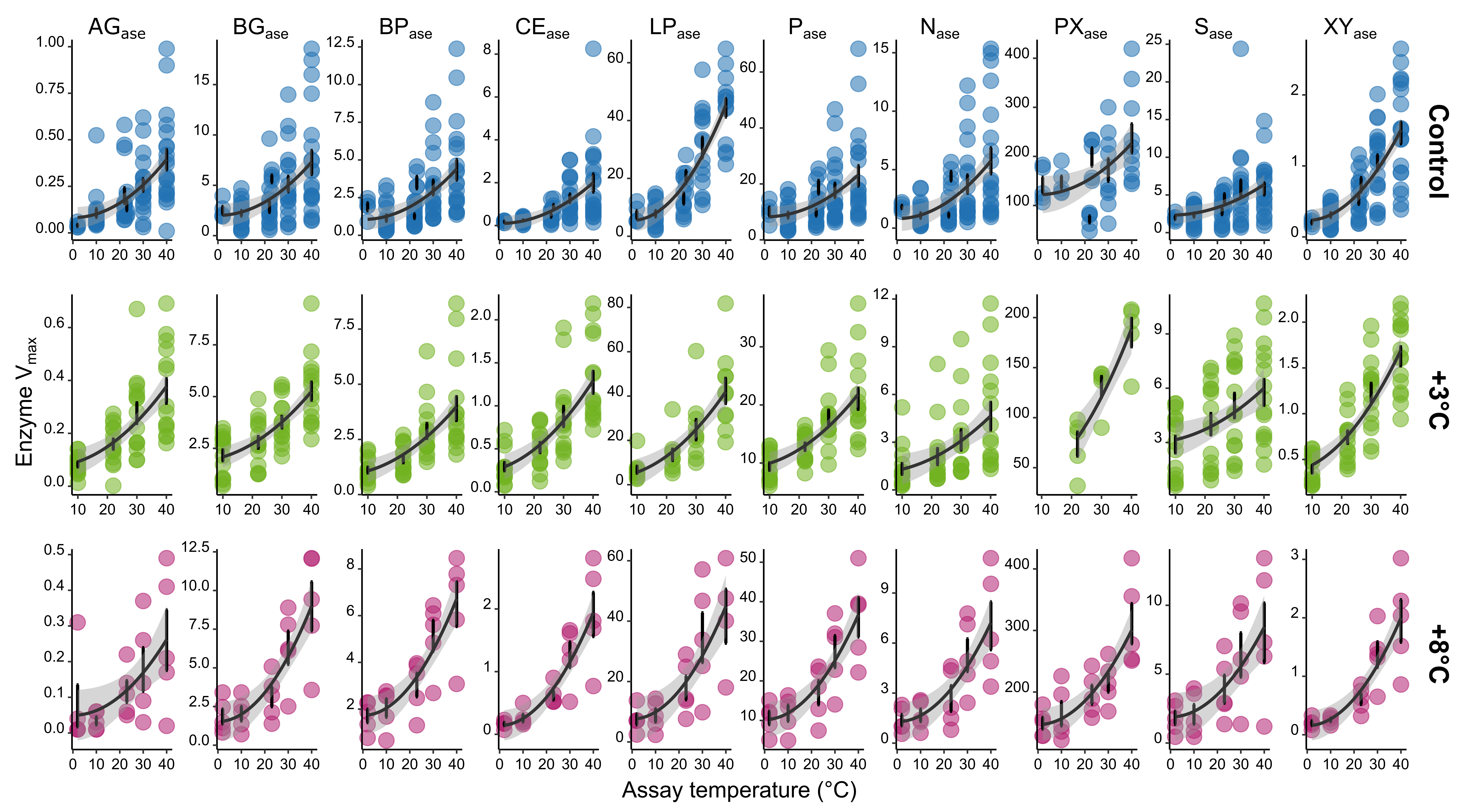
Figure 1
Modifications
Post processing performed in Inkscape. Modifications include rotating combining 16S rRNA and ITS plots, dendrogram branches, changing color palettes, sample and variable renaming, and adding legend.
remove(list =ls())load("include/pub/MAIN/Figure_1.rdata")samp_ps<-c("ssu18_ps_pime")samp_ps_all<-c("ssu18_ps_pime", "ssu18_ps_work")## 1) Get all Class-level Proteobacteria namesfor(iinsamp_ps_all){tmp_name<-purrr::map_chr(i, ~paste0(., "_proteo"))tmp_get<-get(i)tmp_df<-subset_taxa(tmp_get, Phylum=="Proteobacteria")assign(tmp_name, tmp_df)print(tmp_name)tmp_get_taxa<-get_taxa_unique(tmp_df, taxonomic.rank =rank_names(tmp_df)[3], errorIfNULL =TRUE)print(tmp_get_taxa)rm(list =ls(pattern ="tmp_"))#rm(list = ls(pattern = "_proteo"))}## 2) Replace Phylum Proteobacteria with the Class name.for(jinsamp_ps_all){tmp_get<-get(j)tmp_clean<-data.frame(tax_table(tmp_get))for(iin1:nrow(tmp_clean)){if(tmp_clean[i, 2]=="Proteobacteria"&tmp_clean[i, 3]=="Alphaproteobacteria"){phylum<-base::paste("Alphaproteobacteria")tmp_clean[i, 2]<-phylum}elseif(tmp_clean[i, 2]=="Proteobacteria"&tmp_clean[i, 3]=="Gammaproteobacteria"){phylum<-base::paste("Gammaproteobacteria")tmp_clean[i, 2]<-phylum}elseif(tmp_clean[i, 2]=="Proteobacteria"&tmp_clean[i, 3]=="Zetaproteobacteria"){phylum<-base::paste("Zetaproteobacteria")tmp_clean[i, 2]<-phylum}elseif(tmp_clean[i, 2]=="Proteobacteria"&tmp_clean[i, 3]=="p_Proteobacteria"){phylum<-base::paste("p_Proteobacteria")tmp_clean[i, 2]<-phylum}}tax_table(tmp_get)<-as.matrix(tmp_clean)rank_names(tmp_get)assign(j, tmp_get)print(c(j, tmp_get))print(length(get_taxa_unique(tmp_get, taxonomic.rank =rank_names(tmp_get)[2], errorIfNULL =TRUE)))tmp_path<-file.path("include/pub/MAIN/")rm(list =ls(pattern ="tmp_"))}rm(class, order, phylum)# Visualizing DA ASVs in Anvi’o## Here, we combine the results of the ISA and LEfSe analyses with the ## distribution of ASVs across each sample. We are going to do the analysis ## in [anvi’o](https://github.com/merenlab/anvio)---an advanced analysis ## and visualization platform for ‘omics data [@eren2015anvi]---using the ## `anvi-interactive` command. Anvi’o likes databases but it also ## understands that sometimes you do not have a database. So it offers a ## manual mode. If you type this command you can have a look at the ## relevant pieces we need for the visualization, specifically those under ## the headings MANUAL INPUTS and ADDITIONAL STUFF.## There are also a few files we generate that cannot be loaded directly. ## So, in addition to the files that can be loaded when running the interactive, ## we also have files that must be added to the database created by anvi’o.## 1. View data: in our case, a sample by ASV abundance matrix.## 2. Additional info about each ASV.## 3. Additional info about each sample.## 4. Taxa abundance data for each sample at some rank.## 5. Dendrograms ordering the ASVs and samples (based on view data).## 6. Fasta file of all ASVs in the analysis.## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## Main Steps ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ## ########################################## 1. View data ############################################################## Let’s start with the `-d` or `--view-data` file. This file needs to be ## an ASV by sample matrix of read counts. To simplify the visualization, ## we will use ***all*** ASVs represented by 100 or more total reads, ## including those identified as differentially abundant by the ISA and/or LEfSe. trim_val<-100for(iinsamp_ps){tmp_get<-get(i)tmp_df<-prune_taxa(taxa_sums(tmp_get)>trim_val, tmp_get)tmp_name<-purrr::map_chr(i, ~paste0(., "_trim"))assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_"))}for(iinsamp_ps_all){tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")dir.create(paste(tmp_path, i, sep =""), recursive =TRUE)rm(list =ls(pattern ="tmp_"))}for(iinsamp_ps){tmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_df<-as.data.frame(t(otu_table(tmp_get)))tmp_df<-tmp_df%>%rownames_to_column("Group")tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")write.table(tmp_df, paste(tmp_path, i, "/", "data.txt", sep =""), quote =FALSE, sep ="\t", row.names =FALSE)rm(list =ls(pattern ="tmp_"))}## Or export a table of transformed data.for(iinsamp_ps){tmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_trans<-transform_sample_counts(tmp_get, function(x)1e5*{x/sum(x)})tmp_df<-as.data.frame(t(otu_table(tmp_trans)))tmp_df<-tmp_df%>%rownames_to_column("Group")tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")write.table(tmp_df,paste(tmp_path, i, "/", "data_trans.txt", sep =""), quote =FALSE, sep ="\t", row.names =FALSE)rm(list =ls(pattern ="tmp_"))}########################################## 2. Additional Layers for ASVs ############################################# Next, we need some additional data **about the ASVs** to overlay on the ## visual. This can be anything however what I specifically want are the details ## of the ISA analysis, total reads, and lineage info. ## I warn you; this code will get ugly and I urge you to find a better way.## Start with an ASV + lineage table for the ASVs in the new phyloseq object.for(iinsamp_ps){tmp_get_indval<-get(purrr::map_chr(i, ~paste0(., "_indval_final")))tmp_get_indval<-tmp_get_indval%>%dplyr::rename("Group"="ASV_ID")%>%dplyr::rename("enrich_indval"="group")%>%dplyr::rename("test_indval"="indval")%>%dplyr::rename("pval_indval"="pval")tmp_get_indval<-tmp_get_indval[, 1:5]tmp_get_lefse<-get(purrr::map_chr(i, ~paste0(., "_lefse_final")))tmp_get_lefse<-tmp_get_lefse%>%dplyr::rename("Group"="ASV_ID")%>%dplyr::rename("enrich_lefse"="group")%>%dplyr::rename("test_lefse"="lda")%>%dplyr::rename("pval_lefse"="pval")tmp_get_lefse<-tmp_get_lefse[, 1:4]tmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_otu_df<-as.data.frame(t(otu_table(tmp_get)))tmp_total<-cbind(tmp_otu_df, total_reads =rowSums(tmp_otu_df))tmp_total<-rev(tmp_total)[1]tmp_total<-tmp_total%>%tibble::rownames_to_column("Group")tmp_tax_df<-as.data.frame(tax_table(tmp_get))tmp_tax_df$ASV_SEQ<-NULLtmp_tax_df$ASV_ID<-NULLtmp_tax_df<-tmp_tax_df%>%tibble::rownames_to_column("Group")tmp_add_lay<-dplyr::left_join(tmp_tax_df, tmp_total, by ="Group")%>%dplyr::left_join(., tmp_get_indval, by ="Group")%>%dplyr::left_join(., tmp_get_lefse, by ="Group")tmp_add_lay$ASV_ID<-tmp_add_lay$Grouptmp_add_lay<-tmp_add_lay[, c(1, 16, 8, 12, 9:11, 13:15, 2:7)]tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")write.table(tmp_add_lay,paste(tmp_path, i, "/", "additional_layers.txt", sep =""), quote =FALSE, sep ="\t", row.names =FALSE, na ="")rm(list =ls(pattern ="tmp_"))}########################################## 3. Additional Views for Samples ########################################### Now we want some general data **about the samples** to overlay on the ## visual. Again, this can be anything. How about a table of alpha diversity ## metrics? We actually have such a table that was generated way back up the ## road. Just need to fix the column names.metadata_tab[,c(2:5)]<-list(NULL)for(iinsamp_ps){tmp_get<-get(i)tmp_df<-data.frame(sample_data(tmp_get))tmp_df<-tmp_df[,c(2:9)]tmp_df<-tmp_df%>%tibble::rownames_to_column("id")tmp_df<-tmp_df%>%dplyr::rename("no_asvs"="Observed")tmp_rc<-data.frame(readcount(tmp_get))tmp_rc<-tmp_rc%>%tibble::rownames_to_column("id")tmp_rc<-tmp_rc%>%dplyr::rename("no_reads"=2)tmp_merge<-dplyr::left_join(tmp_df, tmp_rc)tmp_merge<-tmp_merge[, c(1:6,10,7:9)]tmp_final<-dplyr::left_join(tmp_merge, metadata_tab, by =c("id"="Sample_ID"))tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")write.table(tmp_final, paste(tmp_path, i, "/", "additional_views.txt", sep =""), quote =FALSE, sep ="\t", row.names =FALSE)rm(list =ls(pattern ="tmp_"))}########################################## 4. Taxon rank abundance by sample ########################################## Turned out this was a little tricky to figure out, but thanks to a ## [little nifty block of code](https://github.com/joey711/phyloseq/issues/418#issuecomment-262637034) ## written by [guoyanzhao](https://github.com/guoyanzhao) on the phyloseq ## Issues forum, it was a piece of cake. The code can be altered to take any ## rank. See the post for an explanation.## Anyway, the goal is to sum each taxon at some rank and present that as a ## bar chart for each sample in the visualization. Anvi'o has a specific format ## it needs where each row is a sample and each column is a taxon. Taxa names ## need the prefix `t_<RANK>!`. For example, `t_class!` should be added for ## Class rank.pick_rank<-"Phylum"pick_rank_l<-"phylum"for(iinsamp_ps_all){# Make the tabletmp_get<-get(i)tmp_glom<-tax_glom(tmp_get, taxrank =pick_rank)tmp_melt<-psmelt(tmp_glom)tmp_melt[[pick_rank]]<-as.character(tmp_melt[[pick_rank]])tmp_abund<-aggregate(Abundance~Sample+tmp_melt[[pick_rank]], tmp_melt, FUN =sum)colnames(tmp_abund)[2]<-"tax_rank"library(reshape2)tmp_abund<-as.data.frame(reshape::cast(tmp_abund, Sample~tax_rank))tmp_abund<-tibble::remove_rownames(tmp_abund)tmp_abund<-tibble::column_to_rownames(tmp_abund, "Sample")# Reorder table column by sumtmp_layers<-tmp_abund[,names(sort(colSums(tmp_abund), decreasing =TRUE))]# Add the prefixtmp_layers<-tmp_layers%>%dplyr::rename_all(function(x)paste0("t_", pick_rank_l,"!", x))tmp_layers<-tibble::rownames_to_column(tmp_layers, "taxon")# save the dataframetmp_name<-paste(i, "_taxa", sep ="")assign(tmp_name, tmp_layers)rm(list =ls(pattern ="tmp_"))}## REORDER TAXAssu18_ps_work_taxa<-dplyr::relocate(ssu18_ps_work_taxa, c("t_phylum!Alphaproteobacteria", "t_phylum!Gammaproteobacteria", "t_phylum!Acidobacteriota", "t_phylum!Actinobacteriota", "t_phylum!Bacteroidota", "t_phylum!Firmicutes","t_phylum!Myxococcota","t_phylum!Verrucomicrobiota","t_phylum!Myxococcota","t_phylum!Chloroflexi","t_phylum!Planctomycetota","t_phylum!Methylomirabilota","t_phylum!Crenarchaeota"), .after ="taxon")write.table(ssu18_ps_work_taxa, "include/pub/MAIN/anvio/ssu/ssu18_ps_work/tax_layers_mod.txt", quote =FALSE, sep ="\t", row.names =FALSE)ssu18_ps_pime_taxa<-dplyr::relocate(ssu18_ps_pime_taxa, c("t_phylum!Alphaproteobacteria", "t_phylum!Gammaproteobacteria", "t_phylum!Acidobacteriota", "t_phylum!Actinobacteriota", "t_phylum!Bacteroidota", "t_phylum!Firmicutes","t_phylum!Myxococcota","t_phylum!Verrucomicrobiota","t_phylum!Myxococcota","t_phylum!Chloroflexi","t_phylum!Planctomycetota","t_phylum!Methylomirabilota","t_phylum!Crenarchaeota"), .after ="taxon")write.table(ssu18_ps_pime_taxa, "include/pub/MAIN/anvio/ssu/ssu18_ps_pime/tax_layers_mod.txt", quote =FALSE, sep ="\t", row.names =FALSE)rm(list =ls(pattern ="tmp_"))################################# 5. Construct Dendrograms ################################# The last piece we need is to generate dendrograms that order the ASVs ## by their distribution in the samples and the samples by their ASV composition. ## For this task we will use anvi'o.## The first command reads the view data we generated above and uses ## Euclidean distance and Ward linkage for hierarchical clustering of the ASVs. ## The second command transposes the view data table and then does the same for ## the samples. There are several distance metrics and linkage methods available. ## See the help menu for the command by typing `anvi-matrix-to-newick -h`. Boom.#bash_commands <- c() USE this to combine all commands# including in loop creates separate filesfor(iinsamp_ps){bash_commands<-c()tmp_command_asv<-purrr::map_chr(i, ~paste0("anvi-matrix-to-newick data.txt"," --distance euclidean --linkage ward -o ","asv.tre"))bash_commands<-append(bash_commands, tmp_command_asv)tmp_command_samp<-purrr::map_chr(i, ~paste0("anvi-matrix-to-newick data.txt"," --distance braycurtis --linkage complete -o ","sample.tre --transpose"))bash_commands<-append(bash_commands, tmp_command_samp)tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")write(bash_commands, paste(tmp_path, i, "/", "tre.sh", sep =""))rm(list =ls(pattern ="tmp_"))}# FOR TANSFORMED DATAfor(iinsamp_ps){bash_commands<-c()tmp_command_asv<-purrr::map_chr(i, ~paste0("anvi-matrix-to-newick data_trans.txt"," --distance euclidean --linkage ward -o ","asv_trans.tre"))bash_commands<-append(bash_commands, tmp_command_asv)tmp_command_samp<-purrr::map_chr(i, ~paste0("anvi-matrix-to-newick data_trans.txt"," --distance braycurtis --linkage complete -o ","sample_trans.tre --transpose"))bash_commands<-append(bash_commands, tmp_command_samp)tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")write(bash_commands, paste(tmp_path, i, "/", "tre_transformed.sh", sep =""))rm(list =ls(pattern ="tmp_"))}
This code is run in anvi’o via a bash script
# The following commands NEED to run in anvio from base dircd include/pub/MAIN/anvio/ssucd ssu18_ps_pimebash tre.shbash tre_transformed.shcd ../
This code is run in R
## Alternatively, we can generate dendrograms using `phyloseq::distance` and `hclust`. pick_dist<-"bray"pick_clust<-"complete"for(iinsamp_ps){# Make the tabletmp_get<-get(i)tmp_dist<-phyloseq::distance(physeq =tmp_get, method =pick_dist, type ="sample")tmp_dend<-hclust(tmp_dist, method =pick_clust)plot(tmp_dend, hang =-1)tmp_tree<-as.phylo(tmp_dend)tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")write.tree(phy =tmp_tree, file =paste(tmp_path, i, "/", "sample_", pick_dist, "_", pick_clust, ".tre", sep =""))rm(list =ls(pattern ="tmp_"))}pick_dist_asv<-"euclidean"pick_clust_asv<-"ward"for(iinsamp_ps){# Make the tabletmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_dist<-phyloseq::distance(physeq =tmp_get, method =pick_dist_asv, type ="taxa")tmp_dend<-hclust(tmp_dist, method =pick_clust_asv)plot(tmp_dend, hang =-1)tmp_tree<-as.phylo(tmp_dend)tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")write.tree(phy =tmp_tree, file =paste(tmp_path, i, "/", "asv_", pick_dist_asv, "_", pick_clust_asv, ".tre", sep =""))rm(list =ls(pattern ="tmp_"))}for(iinsamp_ps){tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")tmp_tree<-read_file(paste(tmp_path, i, "/", "sample.tre", sep =""))tmp_tree<-gsub("[\r\n]", "", tmp_tree)tmp_item<-c("bray_complete")tmp_type<-c("newick")tmp_df<-c(tmp_tree)tmp_tab<-data.frame(tmp_item, tmp_type, tmp_df)tmp_tab%>%janitor::remove_empty("rows")colnames(tmp_tab)<-c("item_name", "data_type", "data_value")write.table(tmp_tab, paste(tmp_path, i, "/", "sample.tre", sep =""), sep ="\t", quote =FALSE, row.names =FALSE, na ="")rm(list =ls(pattern ="tmp_"))}for(iinsamp_ps){tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")tmp_tree<-read_file(paste(tmp_path, i, "/","sample_trans.tre", sep =""))tmp_tree<-gsub("[\r\n]", "", tmp_tree)tmp_item<-c("bray_complete")tmp_type<-c("newick")tmp_df<-c(tmp_tree)tmp_tab<-data.frame(tmp_item, tmp_type, tmp_df)library(janitor)tmp_tab%>%janitor::remove_empty("rows")colnames(tmp_tab)<-c("item_name", "data_type", "data_value")write.table(tmp_tab, paste(tmp_path, i, "/", "sample_trans.tre", sep =""), sep ="\t", quote =FALSE, row.names =FALSE, na ="")rm(list =ls(pattern ="tmp_"))}objects()# FOR HCLUST TREEfor(iinsamp_ps){tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")tmp_tree<-read_file(paste(tmp_path, i, "/", "sample_", pick_dist, "_", pick_clust, ".tre", sep =""))tmp_tree<-gsub("[\r\n]", "", tmp_tree)tmp_item<-c(paste(pick_dist, "_", pick_clust, "_hclust", sep =""))tmp_type<-c("newick")tmp_df<-c(tmp_tree)tmp_tab<-data.frame(tmp_item, tmp_type, tmp_df)library(janitor)tmp_tab%>%janitor::remove_empty("rows")colnames(tmp_tab)<-c("item_name", "data_type", "data_value")write.table(tmp_tab, paste(tmp_path, i, "/", "sample_", pick_dist, "_", pick_clust, ".tre", sep =""), sep ="\t", quote =FALSE, row.names =FALSE, na ="")rm(list =ls(pattern ="tmp_"))}################################# 6. Make a fasta file ##################################### We don't need to add a fasta file, but it is a nice way to keep ## everything in one place. Plus, you can do BLAST searches directly ## in the interface by right clicking on the ASV of interest, so it is nice ## to have the sequences.for(iinsamp_ps){tmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_tab<-tax_table(tmp_get)tmp_tab<-tmp_tab[, 7]tmp_df<-data.frame(row.names(tmp_tab), tmp_tab)colnames(tmp_df)<-c("ASV_ID", "ASV_SEQ")tmp_df$ASV_ID<-sub("^", ">", tmp_df$ASV_ID)tmp_path<-file.path("include/pub/MAIN/anvio/ssu/")write.table(tmp_df, paste(tmp_path, i, "/", i, ".fasta", sep =""), sep ="\n", col.names =FALSE, row.names =FALSE, quote =FALSE, fileEncoding ="UTF-8")rm(list =ls(pattern ="tmp_"))}
This code is run in anvi’o
## Building the Profile Database## Time to put all of these pieces together. This gets a little tricky since ## we do not have a database to start with because some of these files can be ## loaded directly in the interface but some need to be added to a database. ## When we fire up the interactive in `--manual` mode, we ***must*** give ## anvi'o the name of a database and it will *create* that database for us. ## Then we can shut down the interactive, add the necessary data files, ## and start back up.cd ssu18_ps_pimeanvi-interactive--view-data data.txt \--tree asv.tre \--additional-layers additional_layers.txt \--profile-db profile.db \--manual## Now we have a new profile database that we can add the sample metadata ## (`additional_layers.txt`) and the sample dendrogram (sample.tre) using the ## command `anvi-import-misc-data`. These commands add the table to the new ## `profile.db`. First, kill the interactive.anvi-import-misc-data additional_views.txt \--pan-or-profile-db profile.db \--target-data-table layersanvi-import-misc-data sample.tre \--pan-or-profile-db profile.db \--target-data-table layer_orders## One last this is to get the table with the taxonomy total by sample ## (`tax_layers.txt`) into the profile database. We will run the same command ## we just used.anvi-import-misc-data ../ssu18_ps_work/tax_layers_mod.txt \--pan-or-profile-db profile.db \--target-data-table layers## In fact, we could just as easily append the taxonomy total data onto the ## `additional_layers.txt` and import in one command. But we didn't.## Interactive Interface## With a populated database in hand, we can now begin modifying the ## visual by running the interactive command again.anvi-interactive--view-data data.txt \--tree asv.tre \--additional-layers additional_layers.txt \--profile-db profile.db--fasta-file ssu18_ps_pime.fasta \--manual
This code is run in R
## The ITS version of the anvi'o workflow is basically a carbon copy of the ## workflow presented above. It is included here for posterity.## 1. View data: in our case, a sample by ASV abundance matrix.## 2. Additional info about each ASV.## 3. Additional info about each sample.## 4. Taxa abundance data for each sample at some rank.## 5. Dendrograms ordering the ASVs and samples (based on view data).## 6. Fasta file of all ASVs in the analysis.### Main steps########################################## 1. View data ############################################################## Let’s start with the `-d` or `--view-data` file. This file needs to be ## an ASV by sample matrix of read counts. To simplify the visualization, ## we will use ***all*** ASVs represented by 100 or more total reads, ## including those identified as differentially abundant by the ISA and/or LEfSe. samp_ps<-c("its18_ps_pime")samp_ps_all<-c("its18_ps_pime", "its18_ps_work")trim_val<-50for(iinsamp_ps){tmp_get<-get(i)tmp_df<-prune_taxa(taxa_sums(tmp_get)>trim_val, tmp_get)tmp_name<-purrr::map_chr(i, ~paste0(., "_trim"))assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_"))}for(iinsamp_ps_all){tmp_path<-file.path("include/pub/MAIN/anvio/its/")dir.create(paste(tmp_path, i, sep =""), recursive =TRUE)rm(list =ls(pattern ="tmp_"))}for(iinsamp_ps){tmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_df<-as.data.frame(t(otu_table(tmp_get)))tmp_df<-tmp_df%>%rownames_to_column("Group")tmp_path<-file.path("include/pub/MAIN/anvio/its/")write.table(tmp_df, paste(tmp_path, i, "/", "data.txt", sep =""), quote =FALSE, sep ="\t", row.names =FALSE)rm(list =ls(pattern ="tmp_"))}## Or export a table of transformed data.for(iinsamp_ps){tmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_trans<-transform_sample_counts(tmp_get, function(x)1e5*{x/sum(x)})tmp_df<-as.data.frame(t(otu_table(tmp_trans)))tmp_df<-tmp_df%>%rownames_to_column("Group")tmp_path<-file.path("include/pub/MAIN/anvio/its/")write.table(tmp_df,paste(tmp_path, i, "/", "data_trans.txt", sep =""), quote =FALSE, sep ="\t", row.names =FALSE)rm(list =ls(pattern ="tmp_"))}########################################## 2. Additional Layers for ASVs ############################################# Next, we need some additional data **about the ASVs** to overlay on the ## visual. This can be anything however what I specifically want are the ## details of the ISA analysis, total reads, and lineage info. I warn you; ## this code will get ugly and I urge you to find a better way.## Start with an ASV + lineage table for the ASVs in the new phyloseq object.for(iinsamp_ps){tmp_get_indval<-get(purrr::map_chr(i, ~paste0(., "_indval_final")))tmp_get_indval<-tmp_get_indval%>%dplyr::rename("Group"="ASV_ID")%>%dplyr::rename("enrich_indval"="group")%>%dplyr::rename("test_indval"="indval")%>%dplyr::rename("pval_indval"="pval")tmp_get_indval<-tmp_get_indval[, 1:5]tmp_get_lefse<-get(purrr::map_chr(i, ~paste0(., "_lefse_final")))tmp_get_lefse<-tmp_get_lefse%>%dplyr::rename("Group"="ASV_ID")%>%dplyr::rename("enrich_lefse"="group")%>%dplyr::rename("test_lefse"="lda")%>%dplyr::rename("pval_lefse"="pval")tmp_get_lefse<-tmp_get_lefse[, 1:4]tmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_otu_df<-as.data.frame(t(otu_table(tmp_get)))tmp_total<-cbind(tmp_otu_df, total_reads =rowSums(tmp_otu_df))tmp_total<-rev(tmp_total)[1]tmp_total<-tmp_total%>%tibble::rownames_to_column("Group")tmp_tax_df<-as.data.frame(tax_table(tmp_get))tmp_tax_df$ASV_SEQ<-NULLtmp_tax_df$ASV_ID<-NULLtmp_tax_df<-tmp_tax_df%>%tibble::rownames_to_column("Group")tmp_add_lay<-dplyr::left_join(tmp_tax_df, tmp_total, by ="Group")%>%dplyr::left_join(., tmp_get_indval, by ="Group")%>%dplyr::left_join(., tmp_get_lefse, by ="Group")tmp_add_lay$ASV_ID<-tmp_add_lay$Grouptmp_add_lay<-tmp_add_lay[, c(1, 16, 8, 12, 9:11, 13:15, 2:7)]tmp_path<-file.path("include/pub/MAIN/anvio/its/")write.table(tmp_add_lay,paste(tmp_path, i, "/", "additional_layers.txt", sep =""), quote =FALSE, sep ="\t", row.names =FALSE, na ="")rm(list =ls(pattern ="tmp_"))}########################################## 3. Additional Views for Samples ########################################### Now we want some general data **about the samples** to overlay on the visual. ## Again, this can be anything. How about a table of alpha diversity metrics? ## We actually have such a table that was generated way back up the road. ## Just need to fix the column names.metadata_tab<-read.table("files/metadata/tables/metadata.txt", header =TRUE)tmp_x<-readRDS("files/alpha/rdata/its18_ps_pime.rds")data.frame(sample_data(tmp_x))metadata_tab[,c(2:5)]<-list(NULL)for(iinsamp_ps){tmp_get<-get(i)tmp_df<-data.frame(sample_data(tmp_get))tmp_df<-tmp_df[,c(2:9)]tmp_df<-tmp_df%>%tibble::rownames_to_column("id")tmp_df<-tmp_df%>%dplyr::rename("no_asvs"="Observed")tmp_rc<-data.frame(readcount(tmp_get))tmp_rc<-tmp_rc%>%tibble::rownames_to_column("id")tmp_rc<-tmp_rc%>%dplyr::rename("no_reads"=2)#identical(tmp_df$id, tmp_rc$id)tmp_merge<-dplyr::left_join(tmp_df, tmp_rc)tmp_merge<-tmp_merge[, c(1:6,10,7:9)]tmp_final<-dplyr::left_join(tmp_merge, metadata_tab, by =c("id"="Sample_ID"))tmp_path<-file.path("include/pub/MAIN/anvio/its/")write.table(tmp_final, paste(tmp_path, i, "/", "additional_views.txt", sep =""), quote =FALSE, sep ="\t", row.names =FALSE)rm(list =ls(pattern ="tmp_"))}rm(metadata_tab)########################################## 4. Taxon rank abundance by sample ########################################## Turned out this was a little tricky to figure out, but thanks to a ## [little nifty block of code](https://github.com/joey711/phyloseq/iitses/418#iitsecomment-262637034) ## written by [guoyanzhao](https://github.com/guoyanzhao) on the phyloseq ## Issues forum, it was a piece of cake. The code can be altered to take any ## rank. See the post for an explanation.## Anyway, the goal is to sum each taxon at some rank and present that as a ## bar chart for each sample in the visualization. Anvi'o has a specific ## format it needs where each row is a sample and each column is a taxon. ## Taxa names need the prefix `t_<RANK>!`. For example, `t_class!` should be ## added for Class rank.#| code-fold: truepick_rank<-"Order"pick_rank_l<-"order"for(iinsamp_ps_all){# Make the tabletmp_get<-get(i)tmp_glom<-tax_glom(tmp_get, taxrank =pick_rank)tmp_melt<-psmelt(tmp_glom)tmp_melt[[pick_rank]]<-as.character(tmp_melt[[pick_rank]])tmp_abund<-aggregate(Abundance~Sample+tmp_melt[[pick_rank]], tmp_melt, FUN =sum)colnames(tmp_abund)[2]<-"tax_rank"library(reshape2)tmp_abund<-as.data.frame(reshape::cast(tmp_abund, Sample~tax_rank))tmp_abund<-tibble::remove_rownames(tmp_abund)tmp_abund<-tibble::column_to_rownames(tmp_abund, "Sample")# Reorder table column by sumtmp_layers<-tmp_abund[,names(sort(colSums(tmp_abund), decreasing =TRUE))]# Add the prefixtmp_layers<-tmp_layers%>%dplyr::rename_all(function(x)paste0("t_", pick_rank_l,"!", x))tmp_layers<-tibble::rownames_to_column(tmp_layers, "taxon")# save the dataframetmp_name<-paste(i, "_taxa", sep ="")assign(tmp_name, tmp_layers)rm(list =ls(pattern ="tmp_"))}names(its18_ps_work_taxa)names(its18_ps_pime_taxa)## REORDER TAXAits18_ps_work_taxa<-dplyr::relocate(its18_ps_work_taxa, c("t_order!Geastrales", "t_order!Glomerales", "t_order!Helotiales", "t_order!Hypocreales", "t_order!Saccharomycetales", "t_order!Trichosporonales", "t_order!Xylariales", "t_order!Eurotiales", "t_order!Capnodiales", "t_order!Archaeorhizomycetales", "t_order!Agaricales", "t_order!c_Agaricomycetes", "t_order!p_Ascomycota", "t_order!k_Fungi"), .after ="taxon")write.table(its18_ps_work_taxa, "include/pub/MAIN/anvio/its/its18_ps_work/tax_layers_mod.txt", quote =FALSE, sep ="\t", row.names =FALSE)its18_ps_pime_taxa<-dplyr::relocate(its18_ps_pime_taxa, c("t_order!Geastrales", "t_order!Glomerales", "t_order!Helotiales", "t_order!Hypocreales", "t_order!Saccharomycetales", "t_order!Trichosporonales", "t_order!Xylariales", "t_order!Eurotiales", "t_order!Capnodiales", "t_order!Archaeorhizomycetales", "t_order!Agaricales", "t_order!c_Agaricomycetes", "t_order!p_Ascomycota", "t_order!k_Fungi"), .after ="taxon")write.table(its18_ps_pime_taxa, "include/pub/MAIN/anvio/its/its18_ps_pime/tax_layers_mod.txt", quote =FALSE, sep ="\t", row.names =FALSE)rm(list =ls(pattern ="tmp_"))################################# 5. Construct Dendrograms ################################# The last piece we need is to generate dendrograms that order the ASVs ## by their distribution in the samples and the samples by their ## ASV composition. For this task we will use anvi'o.#bash_commands <- c() USE this to combine all commands# including in loop creates separate filesfor(iinsamp_ps){bash_commands<-c()tmp_command_asv<-purrr::map_chr(i, ~paste0("anvi-matrix-to-newick data.txt"," --distance euclidean --linkage ward -o ","asv.tre"))bash_commands<-append(bash_commands, tmp_command_asv)tmp_command_samp<-purrr::map_chr(i, ~paste0("anvi-matrix-to-newick data.txt"," --distance braycurtis --linkage complete -o ","sample.tre --transpose"))bash_commands<-append(bash_commands, tmp_command_samp)tmp_path<-file.path("include/pub/MAIN/anvio/its/")write(bash_commands, paste(tmp_path, i, "/", "tre.sh", sep =""))rm(list =ls(pattern ="tmp_"))}for(iinsamp_ps){bash_commands<-c()tmp_command_asv<-purrr::map_chr(i, ~paste0("anvi-matrix-to-newick data_trans.txt"," --distance euclidean --linkage ward -o ","asv_trans.tre"))bash_commands<-append(bash_commands, tmp_command_asv)tmp_command_samp<-purrr::map_chr(i, ~paste0("anvi-matrix-to-newick data_trans.txt"," --distance braycurtis --linkage complete -o ","sample_trans.tre --transpose"))bash_commands<-append(bash_commands, tmp_command_samp)tmp_path<-file.path("include/pub/MAIN/anvio/its/")write(bash_commands, paste(tmp_path, i, "/", "tre_transformed.sh", sep =""))rm(list =ls(pattern ="tmp_"))}
This code is run in anvi’o via a bash script
#NEED to run in anvio from base dircd its18_ps_pimebash tre.shbash tre_transformed.shcd ../
This code is run in R
## Alternatively, we can generate dendrograms using `phyloseq::distance` and `hclust`. pick_dist<-"bray"pick_clust<-"complete"for(iinsamp_ps){# Make the tabletmp_get<-get(i)tmp_dist<-phyloseq::distance(physeq =tmp_get, method =pick_dist, type ="sample")tmp_dend<-hclust(tmp_dist, method =pick_clust)plot(tmp_dend, hang =-1)tmp_tree<-as.phylo(tmp_dend)tmp_path<-file.path("include/pub/MAIN/anvio/its/")write.tree(phy =tmp_tree, file =paste(tmp_path, i, "/", "sample_", pick_dist, "_", pick_clust, ".tre", sep =""))rm(list =ls(pattern ="tmp_"))}pick_dist_asv<-"euclidean"pick_clust_asv<-"ward"for(iinsamp_ps){# Make the tabletmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_dist<-phyloseq::distance(physeq =tmp_get, method =pick_dist_asv, type ="taxa")tmp_dend<-hclust(tmp_dist, method =pick_clust_asv)plot(tmp_dend, hang =-1)tmp_tree<-as.phylo(tmp_dend)tmp_path<-file.path("include/pub/MAIN/anvio/its/")write.tree(phy =tmp_tree, file =paste(tmp_path, i, "/", "asv_", pick_dist_asv, "_", pick_clust_asv, ".tre", sep =""))rm(list =ls(pattern ="tmp_"))}for(iinsamp_ps){tmp_path<-file.path("include/pub/MAIN/anvio/its/")tmp_tree<-read_file(paste(tmp_path, i, "/", "sample.tre", sep =""))tmp_tree<-gsub("[\r\n]", "", tmp_tree)tmp_item<-c("bray_complete")tmp_type<-c("newick")tmp_df<-c(tmp_tree)tmp_tab<-data.frame(tmp_item, tmp_type, tmp_df)library(janitor)tmp_tab%>%janitor::remove_empty("rows")colnames(tmp_tab)<-c("item_name", "data_type", "data_value")write.table(tmp_tab, paste(tmp_path, i, "/", "sample.tre", sep =""), sep ="\t", quote =FALSE, row.names =FALSE, na ="")rm(list =ls(pattern ="tmp_"))}# FOR TRANSFORMED DATAfor(iinsamp_ps){tmp_path<-file.path("include/pub/MAIN/anvio/its/")tmp_tree<-read_file(paste(tmp_path, i, "/","sample_trans.tre", sep =""))tmp_tree<-gsub("[\r\n]", "", tmp_tree)tmp_item<-c("bray_complete")tmp_type<-c("newick")tmp_df<-c(tmp_tree)tmp_tab<-data.frame(tmp_item, tmp_type, tmp_df)library(janitor)tmp_tab%>%janitor::remove_empty("rows")colnames(tmp_tab)<-c("item_name", "data_type", "data_value")write.table(tmp_tab, paste(tmp_path, i, "/", "sample_trans.tre", sep =""), sep ="\t", quote =FALSE, row.names =FALSE, na ="")rm(list =ls(pattern ="tmp_"))}for(iinsamp_ps){tmp_path<-file.path("include/pub/MAIN/anvio/its/")tmp_tree<-read_file(paste(tmp_path, i, "/", "sample_", pick_dist, "_", pick_clust, ".tre", sep =""))tmp_tree<-gsub("[\r\n]", "", tmp_tree)tmp_item<-c(paste(pick_dist, "_", pick_clust, "_hclust", sep =""))tmp_type<-c("newick")tmp_df<-c(tmp_tree)tmp_tab<-data.frame(tmp_item, tmp_type, tmp_df)library(janitor)tmp_tab%>%janitor::remove_empty("rows")colnames(tmp_tab)<-c("item_name", "data_type", "data_value")write.table(tmp_tab, paste(tmp_path, i, "/", "sample_", pick_dist, "_", pick_clust, ".tre", sep =""), sep ="\t", quote =FALSE, row.names =FALSE, na ="")rm(list =ls(pattern ="tmp_"))}################################# 6. Make a fasta file ##################################### We don't need to add a fasta file, but it is a nice way to keep ## everything in one place. Plus, you can do BLAST searches directly ## in the interface by right clicking on the ASV of interest, so it is nice ## to have the sequences.for(iinsamp_ps){tmp_get<-get(purrr::map_chr(i, ~paste0(., "_trim")))tmp_tab<-tax_table(tmp_get)tmp_tab<-tmp_tab[, 7]tmp_df<-data.frame(row.names(tmp_tab), tmp_tab)colnames(tmp_df)<-c("ASV_ID", "ASV_SEQ")tmp_df$ASV_ID<-sub("^", ">", tmp_df$ASV_ID)tmp_path<-file.path("include/pub/MAIN/anvio/its/")write.table(tmp_df, paste(tmp_path, i, "/", i, ".fasta", sep =""), sep ="\n", col.names =FALSE, row.names =FALSE, quote =FALSE, fileEncoding ="UTF-8")rm(list =ls(pattern ="tmp_"))}
This code is run in anvi’o
### Building the Profile Database## Time to put all of these pieces together. This gets a little tricky ## since we do not have a database to start with because some of these ## files can be loaded directly in the interface but some need to be added ## to a database. When we fire up the interactive in `--manual` mode, we ## ***must*** give anvi'o the name of a database and it will *create* that ## database for us. Then we can shut down the interactive, add the necessary ## data files, and start back up.anvi-interactive--view-data data.txt \--tree asv.tre \--additional-layers additional_layers.txt \--profile-db profile.db \--manual## Now we have a new profile database that we can add the sample metadata ## (`additional_layers.txt`) and the sample dendrogram (sample.tre) using the ## command `anvi-import-misc-data`. These commands add the table to the new ## `profile.db`. First, kill the interactive.anvi-import-misc-data additional_views.txt \--pan-or-profile-db profile.db \--target-data-table layersanvi-import-misc-data sample.tre \--pan-or-profile-db profile.db \--target-data-table layer_orders## One last this is to get the table with the taxonomy total by sample ## (`tax_layers.txt`) into the profile database. We will run the same ## command we just used.anvi-import-misc-data tax_layers.txt \--pan-or-profile-db profile.db \--target-data-table layers## In fact, we could just as easily append the taxonomy total data onto ## the `additional_layers.txt` and import in one command. But we didn't.### Interactive Interface## With a populated database in hand, we can now begin modifying the ## visual by running the interactive command again.anvi-interactive--view-data data.txt \--tree asv.tre \--additional-layers additional_layers.txt \--profile-db profile.db--fasta-file anvio.fasta \--manual
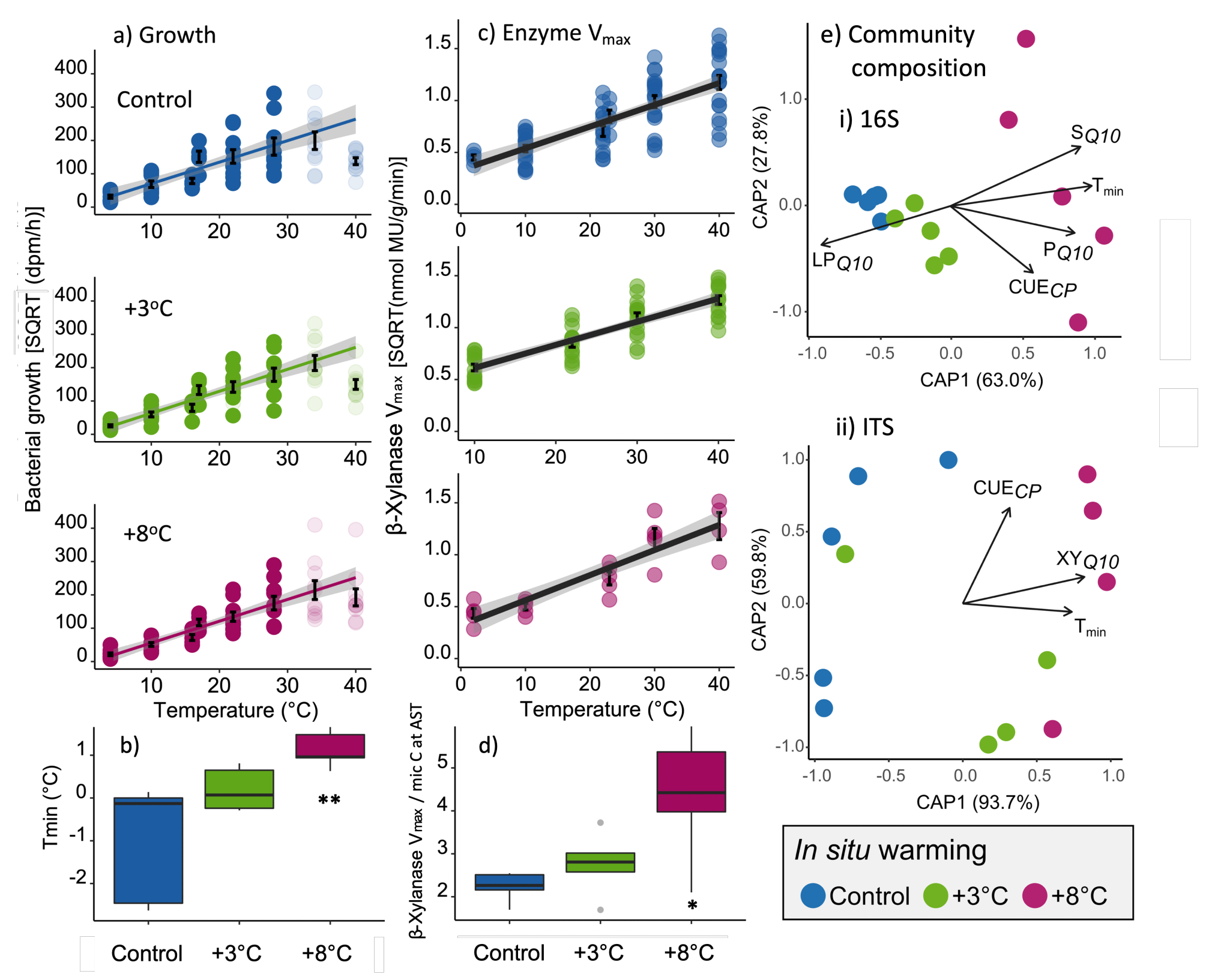
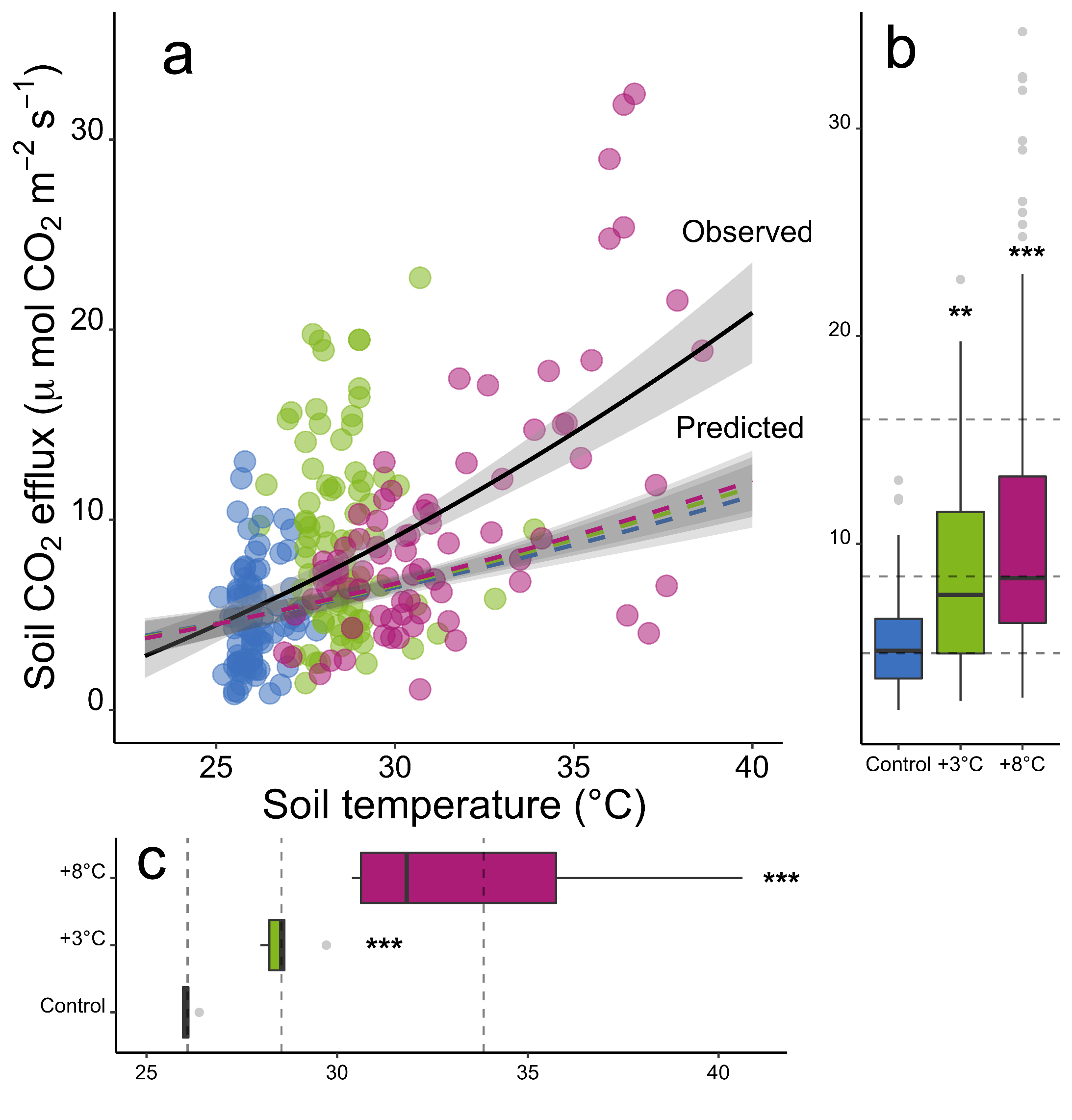
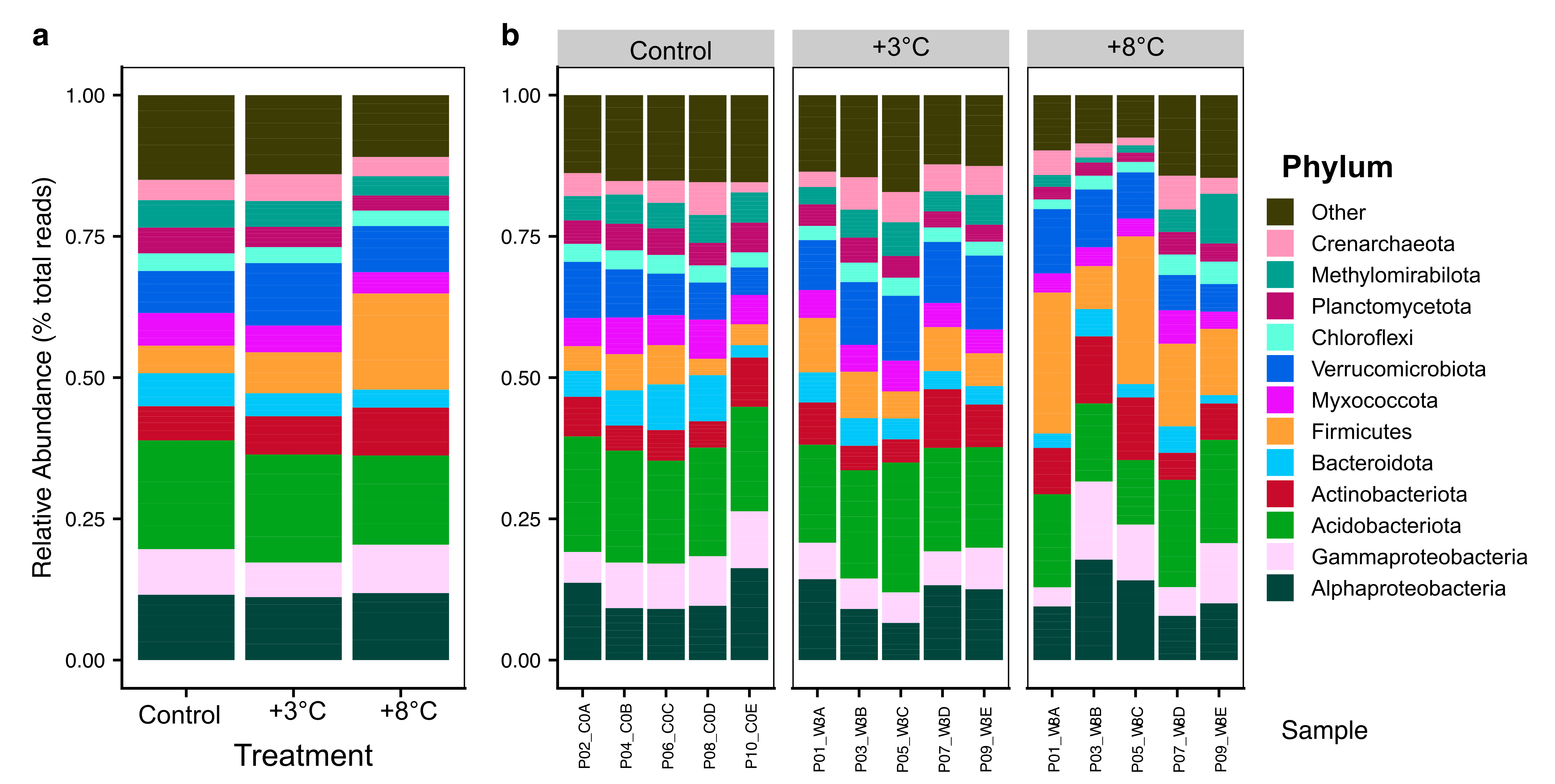
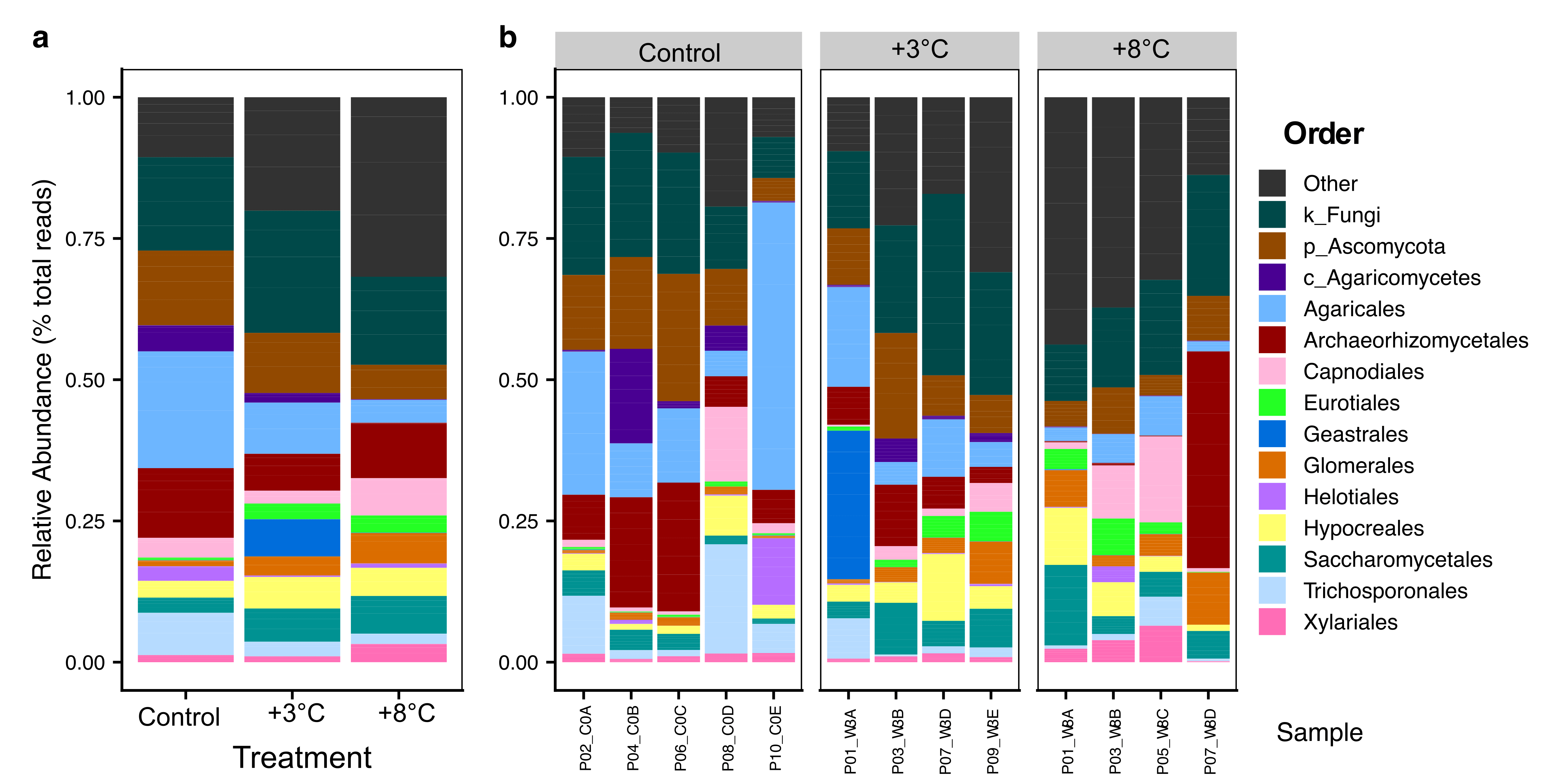
Figure 2
Modifications
Post processing performed in Inkscape. Modifications include sample and variable renaming, and small adjustments in bar height/width.
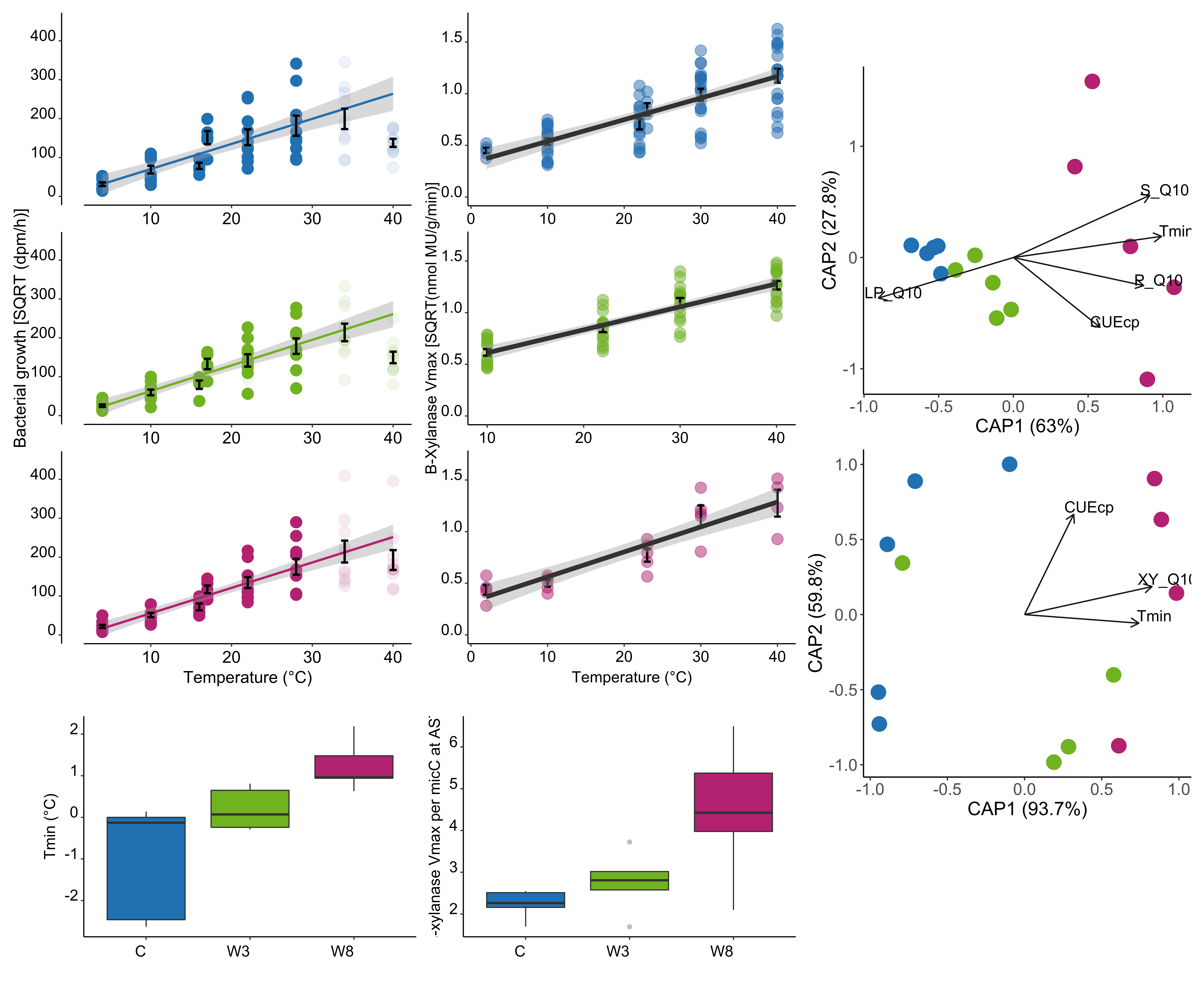
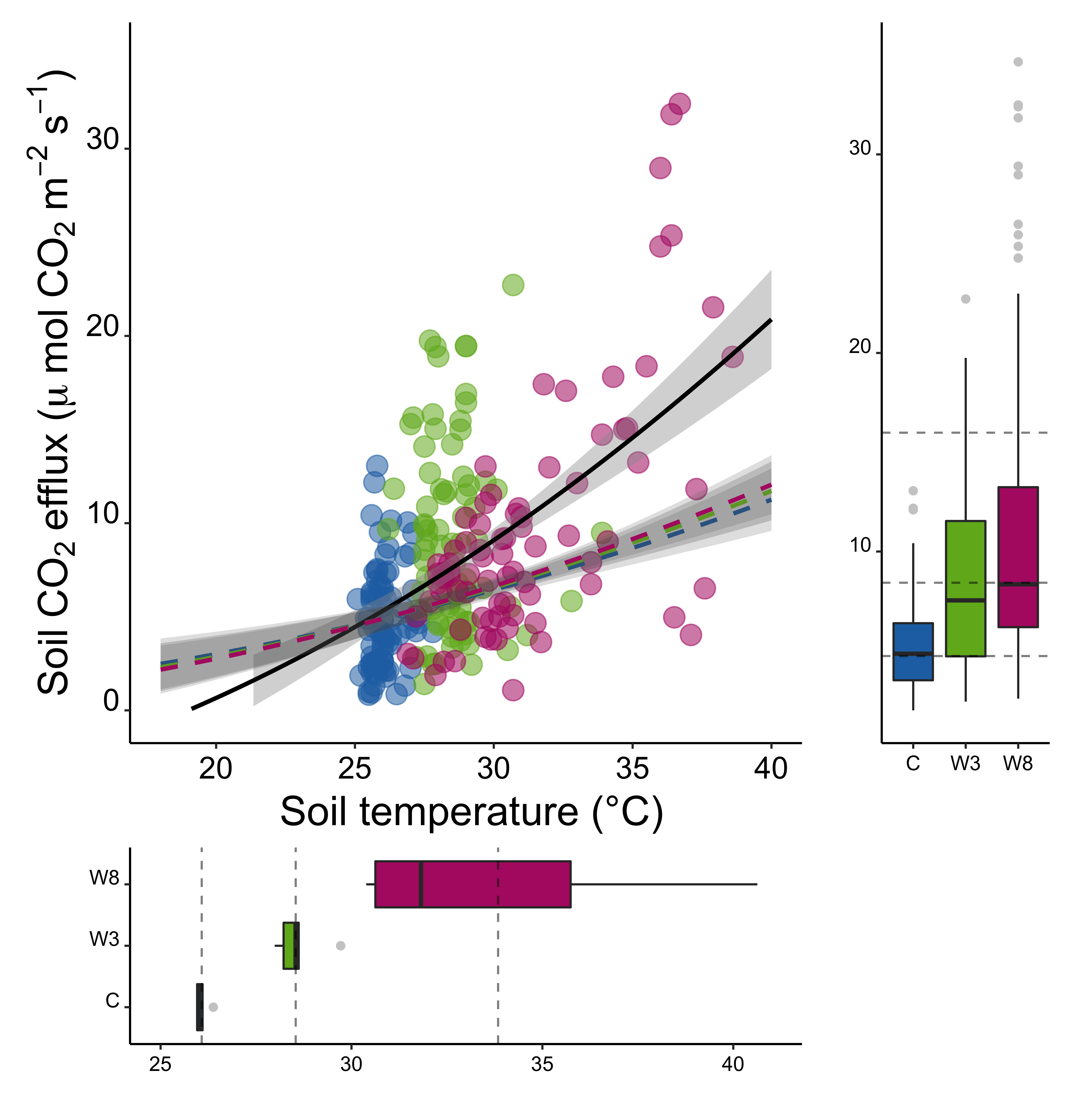
#clear workspacerm(list =ls())# template for blank plotblankPlot<-ggplot()+geom_blank(aes(1, 1))+theme( plot.background =element_blank(), panel.grid.major =element_blank(), panel.grid.minor =element_blank(), panel.border =element_blank(), panel.background =element_blank(), axis.title.x =element_blank(), axis.title.y =element_blank(), axis.text.x =element_blank(), axis.text.y =element_blank(), axis.ticks =element_blank(), axis.line =element_blank())# load data # Predicted fluxes calculated using Tmin model per treatment, ## calculated using CO2 flux at ambient temperature (control plots)#Tmin_CO2pred <-# read.csv("include/pub/MAIN/Fig3_CO2predict.csv", header = T)#CO2c_fullgrad <-# read.csv("include/pub/MAIN/Fig3_CO2observed.csv", header = T)load("include/pub/MAIN/Figure_3.rdata")#determine mean values, measured dataCO2c_fullgrad.means<-plyr::ddply(CO2c_fullgrad,c("Treat", "Plot"),summarise, NCO2 =sum(!is.na(CO2)), CO2 =mean(CO2, na.rm =TRUE), sdCO2 =sd(CO2, na.rm =TRUE), seCO2 =sdCO2/sqrt(NCO2), NTemp =sum(!is.na(Temp)), Temp =mean(Temp, na.rm =TRUE), sdTemp =sd(Temp, na.rm =TRUE), seTemp =sdTemp/sqrt(NTemp))#subsets, predicted dataTmin_CO2pred.C<-subset(Tmin_CO2pred, treat=="C")Tmin_CO2pred.W3<-subset(Tmin_CO2pred, treat=="W3")Tmin_CO2pred.W8<-subset(Tmin_CO2pred, treat=="W8")# data in long format , predicted dataCO2predict.C.long<-Tmin_CO2pred.C%>%gather( key =measure, value =value,-treat,-ID,-depth,-year,-Tmin,-Tambient,-CO2.at.Tmin_SQRT,-CO2.at.Tambient_SQRT,-slope)CO2predict.C.long$measure<-as.factor(CO2predict.C.long$measure)CO2predict.C.long$value<-as.numeric(CO2predict.C.long$value)CO2predict.C.long$measure<-plyr::revalue(CO2predict.C.long$measure,c("CO2_predict29"=29,"CO2_predict34"=34,"CO2.at.Tambient"=25.93395,"CO2.at.Tmin"=0))CO2predict.C.long$measure<-as.numeric(as.character(CO2predict.C.long$measure))CO2predict.C.long$temp.predict.C<-CO2predict.C.long$measureCO2predict.C.long$co2.predict.C<-CO2predict.C.long$value#repeat for W3 adaptationCO2predict.W3.long<-Tmin_CO2pred.W3%>%gather( key =measure, value =value,-treat,-ID,-depth,-year,-Tmin,-Tambient,-CO2.at.Tmin_SQRT,-CO2.at.Tambient_SQRT,-slope)CO2predict.W3.long$measure<-as.factor(CO2predict.W3.long$measure)CO2predict.W3.long$value<-as.numeric(CO2predict.W3.long$value)CO2predict.W3.long$measure<-plyr::revalue(CO2predict.W3.long$measure,c("CO2_predict29"=29,"CO2_predict34"=34,"CO2.at.Tambient"=25.93395,"CO2.at.Tmin"=0))CO2predict.W3.long$measure<-as.numeric(as.character(CO2predict.W3.long$measure))CO2predict.W3.long$temp.predict.W3<-CO2predict.W3.long$measureCO2predict.W3.long$co2.predict.W3<-CO2predict.W3.long$value#repeat for W8 adaptationCO2predict.W8.long<-Tmin_CO2pred.W8%>%gather( key =measure, value =value,-treat,-ID,-depth,-year,-Tmin,-Tambient,-CO2.at.Tmin_SQRT,-CO2.at.Tambient_SQRT,-slope)CO2predict.W8.long$measure<-as.factor(CO2predict.W8.long$measure)CO2predict.W8.long$value<-as.numeric(CO2predict.W8.long$value)CO2predict.W8.long$measure<-plyr::revalue(CO2predict.W8.long$measure,c("CO2_predict29"=29,"CO2_predict34"=34,"CO2.at.Tambient"=25.93395,"CO2.at.Tmin"=0))CO2predict.W8.long$measure<-as.numeric(as.character(CO2predict.W8.long$measure))CO2predict.W8.long$temp.predict.W8<-CO2predict.W8.long$measureCO2predict.W8.long$co2.predict.W8<-CO2predict.W8.long$value#subsetCO2c_fullgrad_sub_full<-subset(CO2c_fullgrad, Treat=="C"|Treat=="W3"|Treat=="W8")CO2c_fullgrad_sub<-subset(CO2c_fullgrad.means, Treat=="C"|Treat=="W3"|Treat=="W8")############ scatter plot CO2 with fit #################################plot.CO2byT_gr<-ggplot()+geom_point(data =CO2c_fullgrad, aes( x =Temp, y =CO2, colour =factor(Treat), size =4, alpha =1))+stat_smooth( data =CO2c_fullgrad,aes(x =Temp, y =CO2, group =1), method ="lm", formula =y~I(x^2), se =T, colour ="black", fullrange =TRUE, linetype =c(1))+scale_colour_manual(values =c("#2271b2","#71b222","#b22271","#b22271","#2271b2","#2271b2"))+scale_fill_manual(values =c("#2271b2","#71b222","#b22271","#b22271","#2271b2","#2271b2"))+ylim(c(0, 35))+xlim(c(18, 40))+ylab(bquote('Soil'~CO[2]~'efflux ('*mu~'mol'~CO[2]~m^-2~s^-1*')'))+xlab(bquote(' Soil temperature (°C)'))+theme_classic()+theme( strip.background =element_blank(), legend.text =element_text(size =15), legend.position ="none", legend.title =element_text(color ="white"), axis.text.x =element_text( colour ="black", size =15, angle =0, hjust =.5, vjust =.5, face ="plain"), axis.text.y =element_text( colour ="black", size =15, angle =0, hjust =1, vjust =0, face ="plain"), axis.title.x =element_text( colour ="black", size =20, angle =0, hjust =.5, vjust =0, face ="plain"), axis.title.y =element_text( colour ="black", size =20, angle =90, hjust =.5, vjust =.5, face ="plain"))############boxplot CO2, meansplot_CO2_box_full<-ggplot(CO2c_fullgrad_sub_full, aes(x =Treat, y =CO2))+geom_boxplot( data =CO2c_fullgrad_sub_full,aes(Treat, CO2, fill =Treat), alpha =1, size =0.5, outlier.colour ="grey80")+scale_colour_manual(values =c("#2271b2","#71b222","#b22271","#b22271","#2271b2","#2271b2"))+scale_fill_manual(values =c("#2271b2","#71b222","#b22271","#b22271","#2271b2","#2271b2"))+ylim(c(2, 35))+geom_hline( yintercept =4.736140 , linetype ="dashed", alpha =0.5, size =0.5)+geom_hline( yintercept =8.427807 , linetype ="dashed", alpha =0.5, size =0.5)+geom_hline( yintercept =15.984643 , linetype ="dashed", alpha =0.5, size =0.5)+ylab(bquote(''))+xlab(bquote(''))+theme_classic()+theme( strip.background =element_blank(), legend.text =element_text(size =15), legend.position ="none", legend.title =element_text(color ="white"), axis.text.x =element_text( colour ="black", size =10, angle =0, hjust =.5, vjust =.5, face ="plain"), axis.text.y =element_text( colour ="black", size =10, angle =0, hjust =1, vjust =0, face ="plain"), axis.title.x =element_text( colour ="black", size =10, angle =0, hjust =.5, vjust =0, face ="plain"), axis.title.y =element_text( colour ="black", size =10, angle =90, hjust =.5, vjust =.5, face ="plain"))############boxplot soilT, meansplot_T_box<-ggplot(CO2c_fullgrad_sub, aes(x =Treat, y =Temp))+geom_boxplot( data =CO2c_fullgrad_sub,aes(Treat, Temp, fill =Treat), alpha =1, size =0.5, outlier.colour ="grey80")+scale_colour_manual(values =c("#2271b2","#71b222","#b22271","#b22271","#2271b2","#2271b2"))+scale_fill_manual(values =c("#2271b2","#71b222","#b22271","#b22271","#2271b2","#2271b2"))+ylim(c(25, 41))+geom_hline( yintercept =26.07722 , linetype ="dashed", alpha =0.5, size =0.5)+geom_hline( yintercept =28.53902 , linetype ="dashed", alpha =0.5, size =0.5)+geom_hline( yintercept =33.84000 , linetype ="dashed", alpha =0.5, size =0.5)+ylab(bquote(''))+xlab(bquote(''))+coord_flip()+theme_classic()+theme( strip.background =element_blank(), legend.text =element_text(size =15), legend.position ="none", legend.title =element_text(color ="white"), axis.text.x =element_text( colour ="black", size =10, angle =0, hjust =.5, vjust =.5, face ="plain"), axis.text.y =element_text( colour ="black", size =10, angle =0, hjust =1, vjust =0, face ="plain"), axis.title.x =element_text( colour ="black", size =10, angle =0, hjust =.5, vjust =0, face ="plain"), axis.title.y =element_text( colour ="black", size =10, angle =90, hjust =.5, vjust =.5, face ="plain"))# overlay fitted prediction plots onto CO2 scatterplot.CO2byT_gr_withpredict<-plot.CO2byT_gr+geom_point( data =CO2predict.C.long,aes(x =temp.predict.C, y =co2.predict.C) , colour ="grey40", fill ="#2271b2", size =2, alpha =0)+stat_smooth( data =CO2predict.C.long,aes(x =temp.predict.C, y =co2.predict.C, group =1), method ="lm", formula =y~I(x^2), se =T, fill ="grey40", colour ="#2271b2", fullrange =TRUE, linetype =c(2), alpha =0.2)+geom_point( data =CO2predict.W3.long,aes(x =temp.predict.W3, y =co2.predict.W3) , colour ="grey40", fill ="#71b222", size =2, alpha =0)+stat_smooth( data =CO2predict.W3.long,aes(x =temp.predict.W3, y =co2.predict.W3, group =1), method ="lm", formula =y~I(x^2), se =T, fill ="grey20", colour ="#71b222", fullrange =TRUE, linetype =c(2), alpha =0.2)+geom_point( data =CO2predict.W8.long,aes(x =temp.predict.W8, y =co2.predict.W8) , colour ="grey40", fill ="#b22271", size =2, alpha =0)+stat_smooth( data =CO2predict.W8.long,aes(x =temp.predict.W8, y =co2.predict.W8, group =1), method ="lm", formula =y~I(x^2), se =T, fill ="grey40", colour ="#b22271", fullrange =TRUE, linetype =c(2), alpha =0.2)# composite plotgridExtra::grid.arrange(plot_T_box, blankPlot, plot.CO2byT_gr_withpredict, plot_CO2_box_full, ncol =2, nrow =2, widths =c(4, 1.4), heights =c(1.4, 4))layout<-c(area(t =1, b =11, l =1, r =4),area(t =12, b =14, l =1, r =4),area(t =1, b =11, l =5, r =5))plot(layout)combo_plot<-plot.CO2byT_gr_withpredict+plot_T_box+plot_CO2_box_full+plot_layout(design =layout)#combo_plot <- # plot.Tmin_forVmax + plot.vmax.XY + plot.Tminxyl + ((ssu18_temp_adapt_plot / its18_temp_adapt_plot)) + # plot_layout(design = layout) ggplot2::ggsave(combo_plot, path ="include/pub/MAIN/", filename ="Figure_3.png", height =19.33, width =18.51, units ='cm', dpi =600, bg ="white")ggplot2::ggsave(combo_plot, path ="include/pub/MAIN/", filename ="Figure_3.pdf", height =19.33, width =18.51, units ='cm', dpi =600, bg ="white")
Extended Data
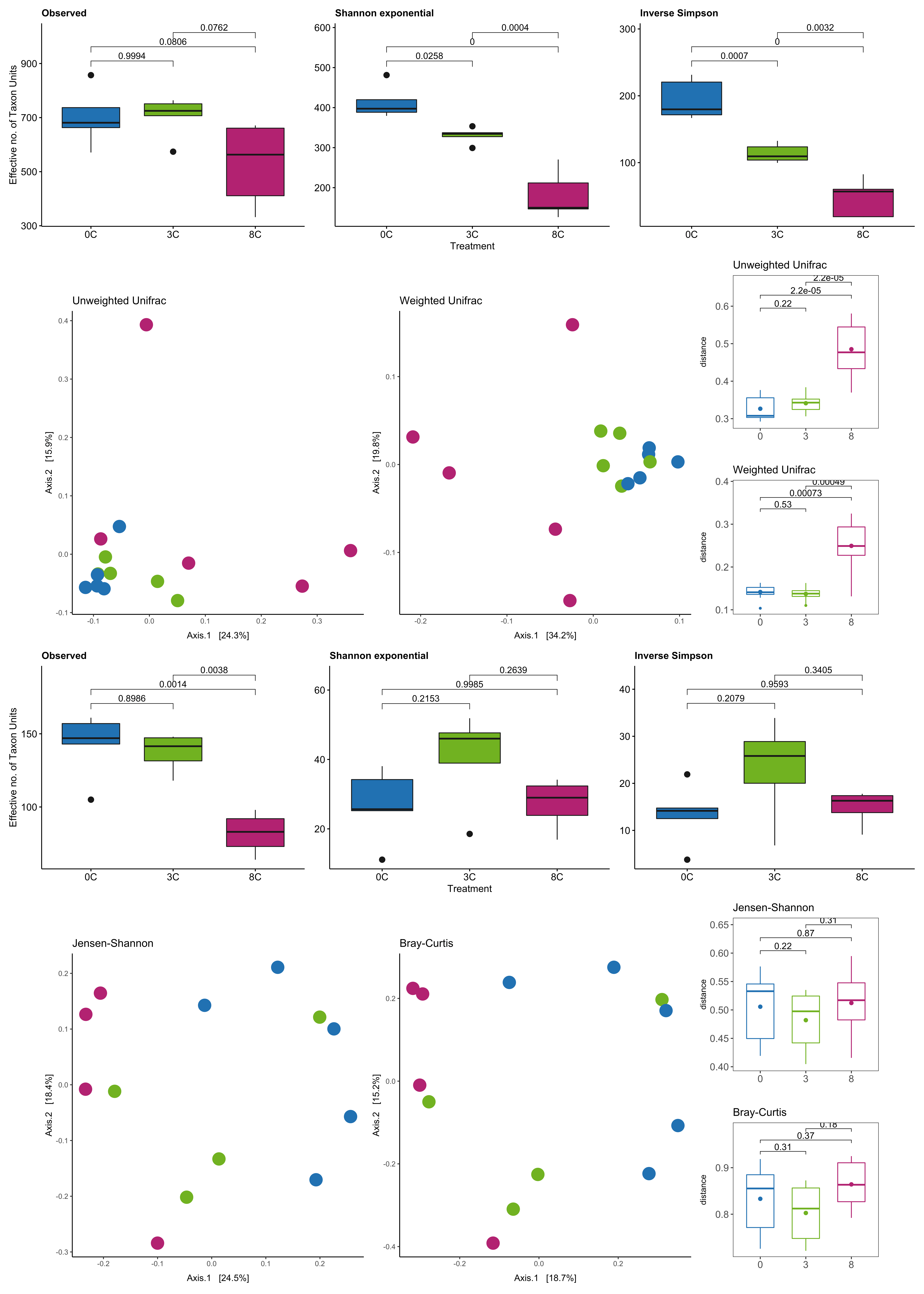
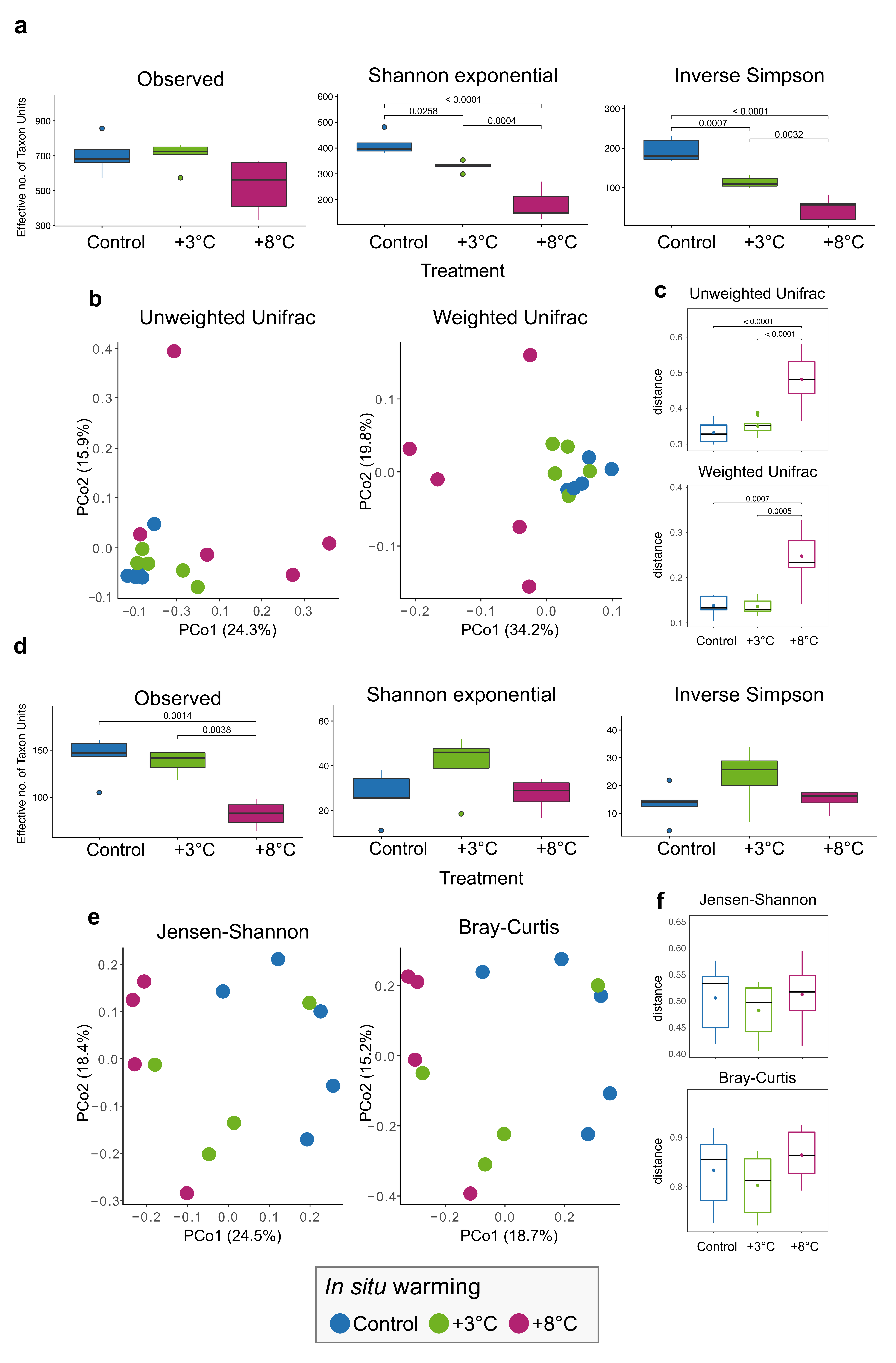
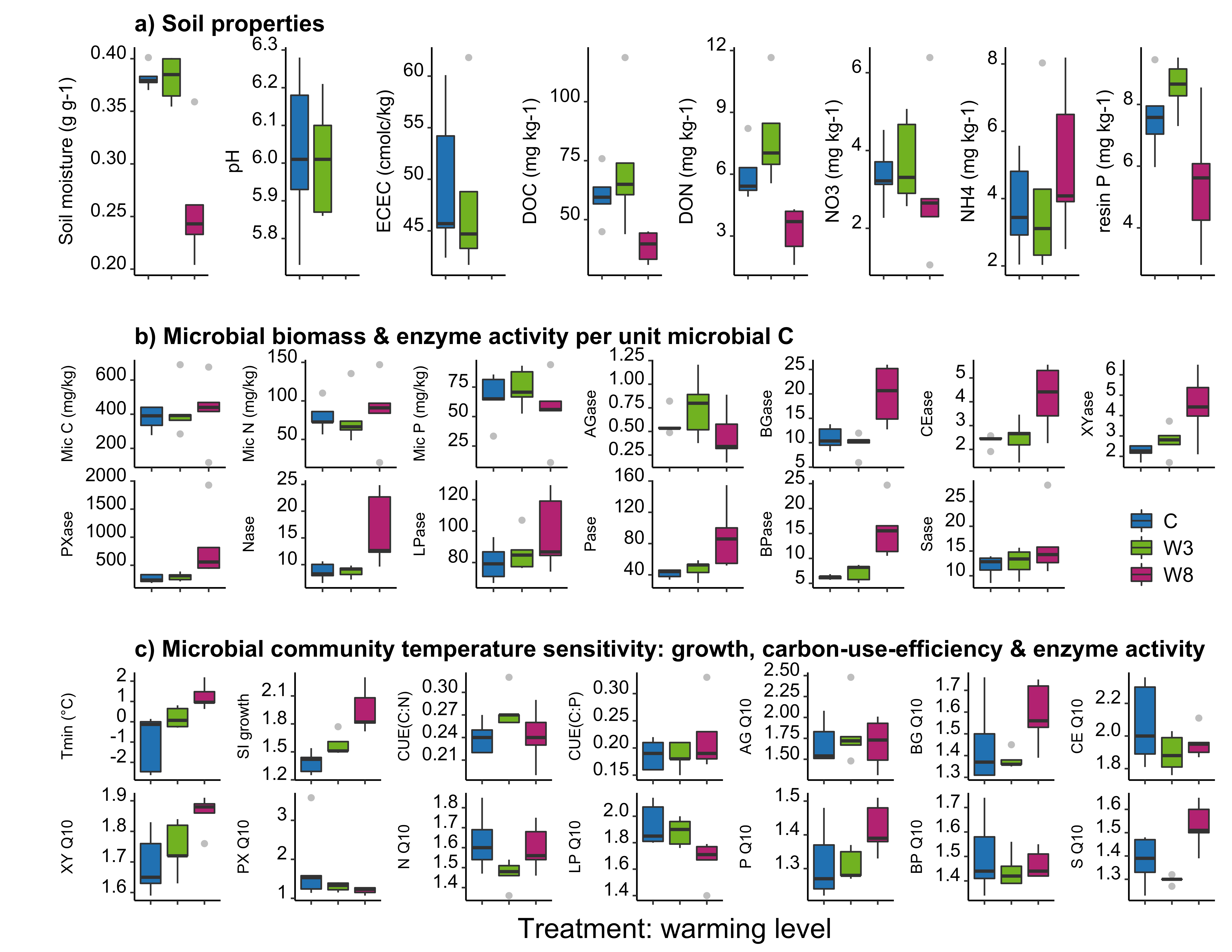
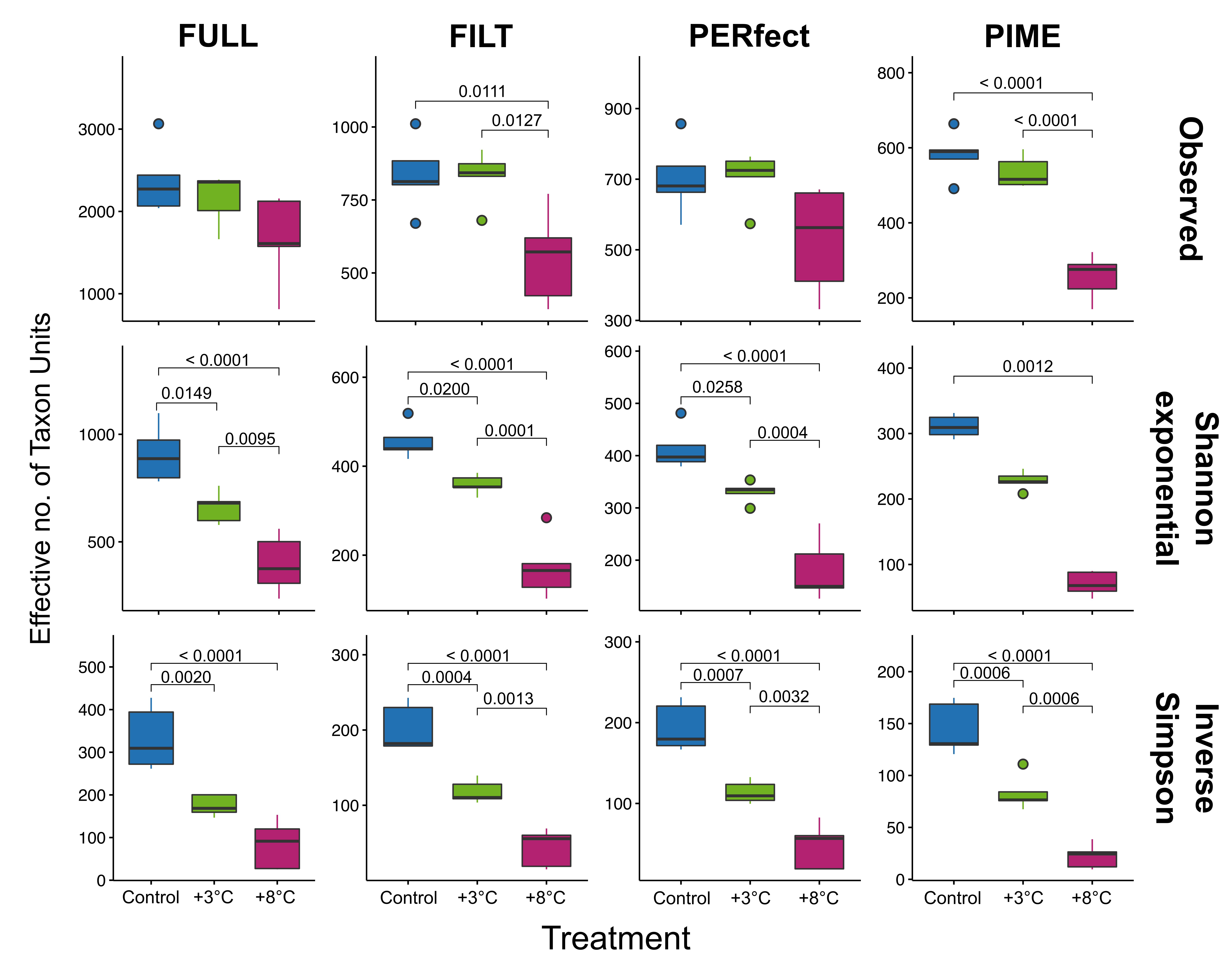
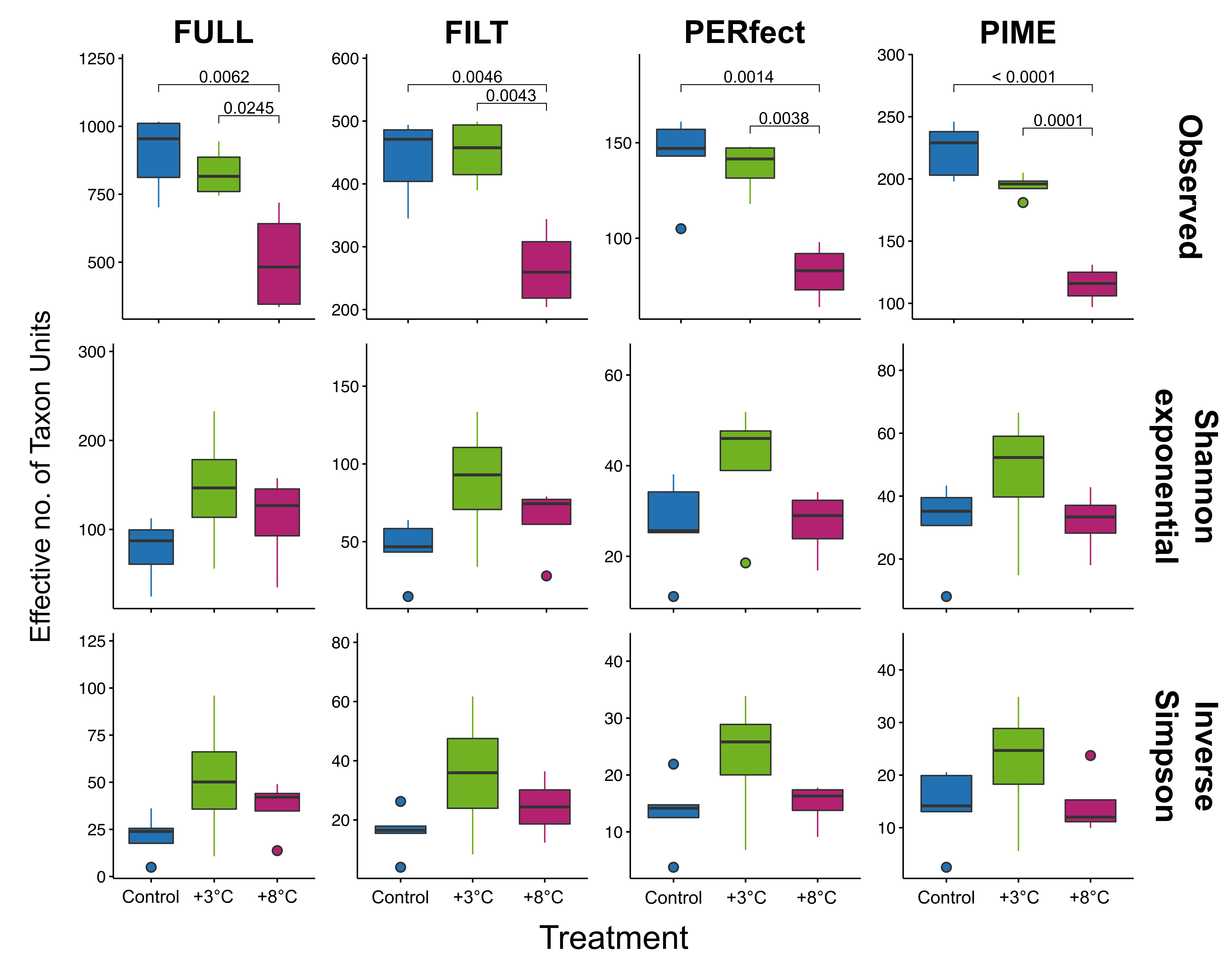
Extended Data Figure 2
Modifications
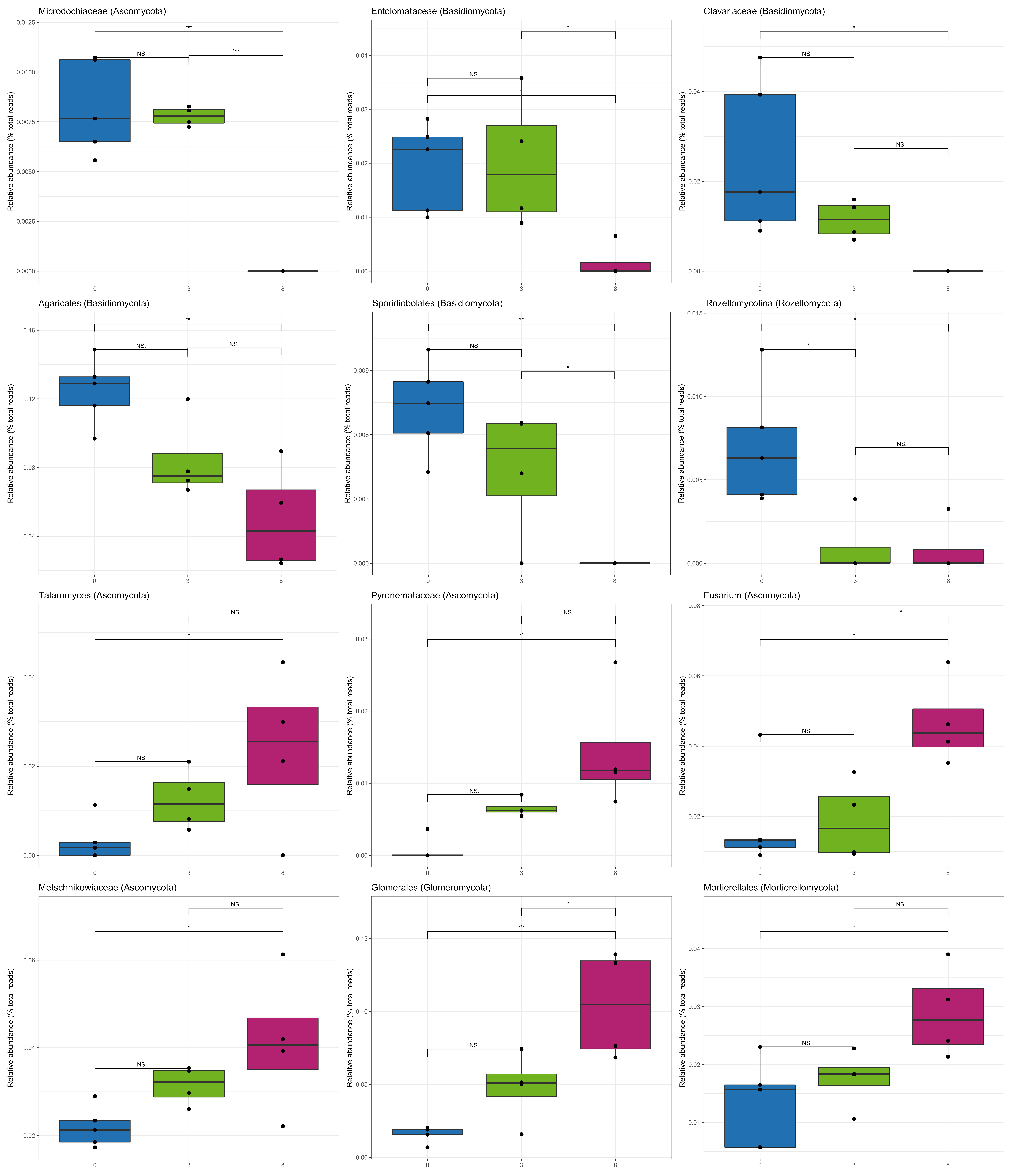
Post processing performed in Inkscape. Modifications include axes and label resizing, removing non-significant (NS) values from plots, changing significant p-values to asterisks (*), creating a legend, and enlarging outlier points.
remove(list =ls())load("include/pub/EXD/EXD_Figure_2/Extended_Data_Figure_2.rdata")############################################################################# 16S rRNA ALPHA DIV PLOTS ##################################################################################tmp_objects<-c("ssu18_ps_perfect")tmp_metric<-data.frame(c("Observed", "Shannon exponential", "Inverse Simpson"))tmp_qvalue<-data.frame(c("0", "1", "2"))qvalue<-c(0,1,2)for(iintmp_objects){tmp_h_pvalue<-c()for(jinqvalue){tmp_name<-purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))tmp_get<-get(tmp_name)$homogeneity.pvaluetmp_h_pvalue<-c(append(tmp_h_pvalue, tmp_get))}tmp_h_pvalue<-data.frame(tmp_h_pvalue)tmp_n_pvalue<-c()for(jinqvalue){tmp_name<-purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))tmp_get<-get(tmp_name)$normality.pvaluetmp_n_pvalue<-c(append(tmp_n_pvalue, tmp_get))}tmp_n_pvalue<-data.frame(tmp_n_pvalue)tmp_method<-c()for(jinqvalue){tmp_name<-purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))tmp_get<-get(tmp_name)$methodtmp_method<-c(append(tmp_method, tmp_get))}tmp_method<-data.frame(tmp_method)tmp_phoc_method<-c()for(jinqvalue){tmp_name<-purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))tmp_get<-get(tmp_name)$posthoc.methodtmp_phoc_method<-c(append(tmp_phoc_method, tmp_get))}tmp_phoc_method<-data.frame(tmp_phoc_method)tmp_df<-dplyr::bind_cols(tmp_metric, tmp_qvalue)%>%dplyr::bind_cols(., tmp_n_pvalue)%>%dplyr::bind_cols(., tmp_h_pvalue)%>%dplyr::bind_cols(., tmp_method)%>%dplyr::bind_cols(., tmp_phoc_method)%>%dplyr::rename("metric"=1, "q-value"=2, "normality p-value"=3, "homogeneity p-value"=4, "method"=5, "posthoc method"=6)tmp_name<-purrr::map_chr(i, ~paste0(i, "_sig_tab"))assign(tmp_name, tmp_df)}## FALSE Kruskal-Wallis Test = its18_pime_q1_adt$test[[3]]## TRUE Tukey post-hoc = its18_ps_work_q0_adt$test[[1]][[1, 5]]tmp_pvalue<-data.frame(c(ssu18_ps_perfect_q0_adt$test[[1]][[1, 5]],ssu18_ps_perfect_q1_adt$test[[1]][[1, 5]],ssu18_ps_perfect_q2_adt$test[[1]][[1, 5]]))ssu18_ps_perfect_sig_tab<-dplyr::bind_cols(ssu18_ps_perfect_sig_tab, tmp_pvalue)%>%dplyr::rename("posthoc p-value"=7)rm(list =ls(pattern ="tmp_"))ssu18_ps_perfect_sig_tab$dataset<-"PERfect"ssu18_ps_perfect_sig_tab$type<-"ASV"ssu18_ps_perfect_sig_tab$lineage<-"No"ssu18_ps_perfect_sig_tab<-ssu18_ps_perfect_sig_tab[,c(8:10,1:7)]ssu18_sig_tab_all<-ssu18_ps_perfect_sig_tab## PostHoc Analyses ## First let's check the results of each posthoc analysis.### Observed (q-value = 0)ssu18_asvr0_lab<-"Observed"ssu18_asvr0_labssu18_ps_perfect_q0_adt$posthoc.methoddata.frame(ssu18_ps_perfect_q0_adt$posthoc)ssu18_asvr1_lab<-"Shannon exponential"ssu18_asvr1_labssu18_ps_perfect_q1_adt$posthoc.methoddata.frame(ssu18_ps_perfect_q1_adt$posthoc)ssu18_asvr2_lab<-"Inverse Simpson"ssu18_asvr2_labssu18_ps_perfect_q2_adt$posthoc.methoddata.frame(ssu18_ps_perfect_q2_adt$posthoc)## Now we can plot the results from the posthoc analyses for each metric ## and data set using the function `div_test_plot_jjs`. ## I modified the original function (`div_test_plot`) to control a ## little of the formatting.## The command is as follows:## div_test_plot(divtest = x, chart = "type", colour = col.pal, ## posthoc = TRUE, threshold = value))## where `x` is the results from the `div_test` function, `"type"` is ## chart type (box, jitter, or violin), `colour` is is a color palette, ## `posthoc` indicates whether to run posthoc pairwise analyses, and `value` ## is the maximum p-value to show in pairwise posthoc results. ## **WARNING** if none of the posthoc results are below the specified ## threshold, the function will throw an error. Therefore, until this is ## fixed, all posthoc values are shown.div_test_plot_jjs<-function(divtest, chart, colour, posthoc, threshold){if(missing(chart)){chart="box"}if(missing(posthoc)){posthoc=FALSE}if((names(divtest)[1]!="data")&(names(divtest)[2]!="normality.pvalue"))stop("The input object does not seem to be a div_test output.")divtestdata<-divtest$datadivtestdata$Group<-as.factor(divtestdata$Group)divtestdata$Group<-factor(divtestdata$Group, levels =as.character(unique(divtestdata$Group)))if(missing(colour)||(length(colour)<divtest$groups)){getPalette<-colorRampPalette(brewer.pal(divtest$groups, "Paired"))colour<-getPalette(divtest$groups)}if(posthoc==TRUE){if(is.na(names(divtest)[7]))stop("The input div_test object does not seem to contain pairwise posthoc data. Re-run div_test() using 'posthoc=TRUE' argument.")if(divtest[7]=="Tukey post-hoc test"){combinations<-matrix(gsub(" $", "", gsub("^ ", "", unlist(strsplit(as.character(rownames(divtest$posthoc)), "-", fixed =TRUE)))), ncol =2, byrow =TRUE)pvalue<-round(divtest$posthoc[, 4], 4)pairwisetable<-as.data.frame(cbind(combinations, pvalue))colnames(pairwisetable)<-c("group1", "group2", "p")}if(divtest[7]=="Dunn test with Benjamini-Hochberg correction"){combinations<-matrix(gsub(" $", "", gsub("^ ", "", unlist(strsplit(as.character(rownames(divtest$posthoc)), "-", fixed =TRUE)))), ncol =2, byrow =TRUE)pvalue<-round(divtest$posthoc[, 3], 4)pairwisetable<-as.data.frame(cbind(combinations, pvalue))colnames(pairwisetable)<-c("group1", "group2", "p")}pairwisetable[, 1]<-as.character(pairwisetable[, 1])pairwisetable[, 2]<-as.character(pairwisetable[, 2])pairwisetable[, 3]<-as.numeric(as.character(pairwisetable[, 3]))if(!missing(threshold)){pairwisetable<-pairwisetable[which(pairwisetable$p<threshold), ]}sortedgroups<-unique(sort(c(pairwisetable$group1, pairwisetable$group2)))datamax<-round(max(divtest$data[which(divtest$data$Group%in%sortedgroups), 3]))datamin<-round(min(divtest$data[which(divtest$data$Group%in%sortedgroups), 3]))datarange<-datamax-dataminby<-datarange*0.1min<-datamaxmax<-min+(by*nrow(pairwisetable))ypos<-seq(min, max, by)[-1]pairwisetable$ypos<-ypos}if(chart=="box"){plot<-ggboxplot(divtestdata, x ="Group", y ="Value", outlier.size =3, color ="Group", fill ="Group", x.text.angle =0)+ylab("Effective no. of Taxon Units")+xlab("Treatment")+#scale_colour_manual(values = scales::alpha(colour, 1)) + scale_colour_manual(values=c("#191919", "#191919", "#191919"))+scale_fill_manual(values =scales::alpha(colour, 1))+scale_linetype_manual()if(posthoc==TRUE){plot<-suppressWarnings(plot+stat_pvalue_manual(pairwisetable, label ="p", y.position ="ypos"))}return(plot)}if(chart=="jitter"){plot<-ggboxplot(divtestdata, x ="Group", y ="Value", color ="Group", add ="jitter", width =0, x.text.angle =45)+ylab("Effective no. of Taxon Units")+xlab("Treatment")+scale_colour_manual(values =scales::alpha(colour, 0))if(posthoc==TRUE){plot<-suppressWarnings(plot+stat_pvalue_manual(pairwisetable, label ="p", y.position ="ypos"))}print(plot)}if(chart=="violin"){plot<-ggviolin(divtestdata, x ="Group", y ="Value", color ="Group", fill ="Group", x.text.angle =45)+ylab("Effective no. of Taxon Units")+xlab("Treatment")+scale_fill_manual(values =scales::alpha(colour, 0.1))+scale_colour_manual(values =scales::alpha(colour, 1))if(posthoc==TRUE){plot<-suppressWarnings(plot+stat_pvalue_manual(pairwisetable, label ="p", y.position ="ypos"))}print(plot)}}swel_col<-c("#2271B2", "#71B222", "#B22271")rm(list =ls(pattern ="_adt_plot"))for(iinobjects(pattern ="_adt")){tmp_name<-purrr::map_chr(i, ~paste0(., "_plot"))tmp_get<-get(i)tmp_df<-div_test_plot_jjs(tmp_get, chart ="box", colour =swel_col, posthoc =TRUE)tmp_df<-ggpar(tmp_df, legend ="none")print(tmp_name)assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_"))}#ssu18_ps_perfect_q0_adt_plot<-ssu18_ps_perfect_q0_adt_plot+labs(y ="Effective no. of Taxon Units", x ="")+ggtitle(ssu18_asvr0_lab)+theme(plot.title =element_text(size =12, face ="bold"))ssu18_ps_perfect_q1_adt_plot<-ssu18_ps_perfect_q1_adt_plot+labs(x ="Treatment")+theme(axis.title.y =element_blank())+ggtitle(ssu18_asvr1_lab)+theme(plot.title =element_text(size =12, face ="bold"))ssu18_ps_perfect_q2_adt_plot<-ssu18_ps_perfect_q2_adt_plot+labs(x ="")+theme(axis.title.y =element_blank())+ggtitle(ssu18_asvr2_lab)+theme(plot.title =element_text(size =12, face ="bold"))ssu18_alph_div_plots_asv<-ggarrange(ssu18_ps_perfect_q0_adt_plot, ssu18_ps_perfect_q1_adt_plot, ssu18_ps_perfect_q2_adt_plot, ncol =3, nrow =1)ssu18_alph_div_plots_asv############################################################################# ITS ALPHA DIV PLOTS #######################################################################################tmp_objects<-c("its18_ps_perfect")tmp_metric<-data.frame(c("Observed", "Shannon exponential", "Inverse Simpson"))tmp_qvalue<-data.frame(c("0", "1", "2"))qvalue<-c(0,1,2)for(iintmp_objects){tmp_h_pvalue<-c()for(jinqvalue){tmp_name<-purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))tmp_get<-get(tmp_name)$homogeneity.pvaluetmp_h_pvalue<-c(append(tmp_h_pvalue, tmp_get))}tmp_h_pvalue<-data.frame(tmp_h_pvalue)tmp_n_pvalue<-c()for(jinqvalue){tmp_name<-purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))tmp_get<-get(tmp_name)$normality.pvaluetmp_n_pvalue<-c(append(tmp_n_pvalue, tmp_get))}tmp_n_pvalue<-data.frame(tmp_n_pvalue)tmp_method<-c()for(jinqvalue){tmp_name<-purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))tmp_get<-get(tmp_name)$methodtmp_method<-c(append(tmp_method, tmp_get))}tmp_method<-data.frame(tmp_method)tmp_phoc_method<-c()for(jinqvalue){tmp_name<-purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))tmp_get<-get(tmp_name)$posthoc.methodtmp_phoc_method<-c(append(tmp_phoc_method, tmp_get))}tmp_phoc_method<-data.frame(tmp_phoc_method)tmp_df<-dplyr::bind_cols(tmp_metric, tmp_qvalue)%>%dplyr::bind_cols(., tmp_n_pvalue)%>%dplyr::bind_cols(., tmp_h_pvalue)%>%dplyr::bind_cols(., tmp_method)%>%dplyr::bind_cols(., tmp_phoc_method)%>%dplyr::rename("metric"=1, "q-value"=2, "normality p-value"=3, "homogeneity p-value"=4, "method"=5, "posthoc method"=6)tmp_name<-purrr::map_chr(i, ~paste0(i, "_sig_tab"))assign(tmp_name, tmp_df)}## FALSE Kruskal-Wallis Test = its18_pime_q1_adt$test[[3]]## TRUE Tukey post-hoc = its18_ps_work_q0_adt$test[[1]][[1, 5]]tmp_pvalue<-data.frame(c(its18_ps_perfect_q0_adt$test[[1]][[1, 5]],its18_ps_perfect_q1_adt$test[[1]][[1, 5]],its18_ps_perfect_q2_adt$test[[1]][[1, 5]]))its18_ps_perfect_sig_tab<-dplyr::bind_cols(its18_ps_perfect_sig_tab, tmp_pvalue)%>%dplyr::rename("posthoc p-value"=7)rm(list =ls(pattern ="tmp_"))its18_ps_perfect_sig_tab$dataset<-"PERfect"its18_ps_perfect_sig_tab$type<-"ASV"its18_ps_perfect_sig_tab$lineage<-"No"its18_ps_perfect_sig_tab<-its18_ps_perfect_sig_tab[,c(8:10,1:7)]its18_sig_tab_all<-its18_ps_perfect_sig_tab## PostHoc Analyses ## First let's check the results of each posthoc analysis.### Observed (q-value = 0)its18_asvr0_lab<-"Observed"its18_asvr0_labits18_ps_perfect_q0_adt$posthoc.methoddata.frame(its18_ps_perfect_q0_adt$posthoc)its18_asvr1_lab<-"Shannon exponential"its18_asvr1_labits18_ps_perfect_q1_adt$posthoc.methoddata.frame(its18_ps_perfect_q1_adt$posthoc)its18_asvr2_lab<-"Inverse Simpson"its18_asvr2_labits18_ps_perfect_q2_adt$posthoc.methoddata.frame(its18_ps_perfect_q2_adt$posthoc)swel_col<-c("#2271B2", "#71B222", "#B22271")rm(list =ls(pattern ="_adt_plot"))for(iinobjects(pattern ="_adt")){tmp_name<-purrr::map_chr(i, ~paste0(., "_plot"))tmp_get<-get(i)tmp_df<-div_test_plot_jjs(tmp_get, chart ="box", colour =swel_col, posthoc =TRUE)tmp_df<-ggpar(tmp_df, legend ="none")print(tmp_name)assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_"))}#its18_ps_perfect_q0_adt_plot<-its18_ps_perfect_q0_adt_plot+labs(y ="Effective no. of Taxon Units", x ="")+ggtitle(its18_asvr0_lab)+theme(plot.title =element_text(size =12, face ="bold"))its18_ps_perfect_q1_adt_plot<-its18_ps_perfect_q1_adt_plot+labs(x ="Treatment")+theme(axis.title.y =element_blank())+ggtitle(its18_asvr1_lab)+theme(plot.title =element_text(size =12, face ="bold"))its18_ps_perfect_q2_adt_plot<-its18_ps_perfect_q2_adt_plot+labs(x ="")+theme(axis.title.y =element_blank())+ggtitle(its18_asvr2_lab)+theme(plot.title =element_text(size =12, face ="bold"))its18_alph_div_plots_asv<-ggarrange(its18_ps_perfect_q0_adt_plot, its18_ps_perfect_q1_adt_plot, its18_ps_perfect_q2_adt_plot, ncol =3, nrow =1)its18_alph_div_plots_asv
Beta diversity plot code
Access the code for Extended Data Figure 2 b, c, e, & f
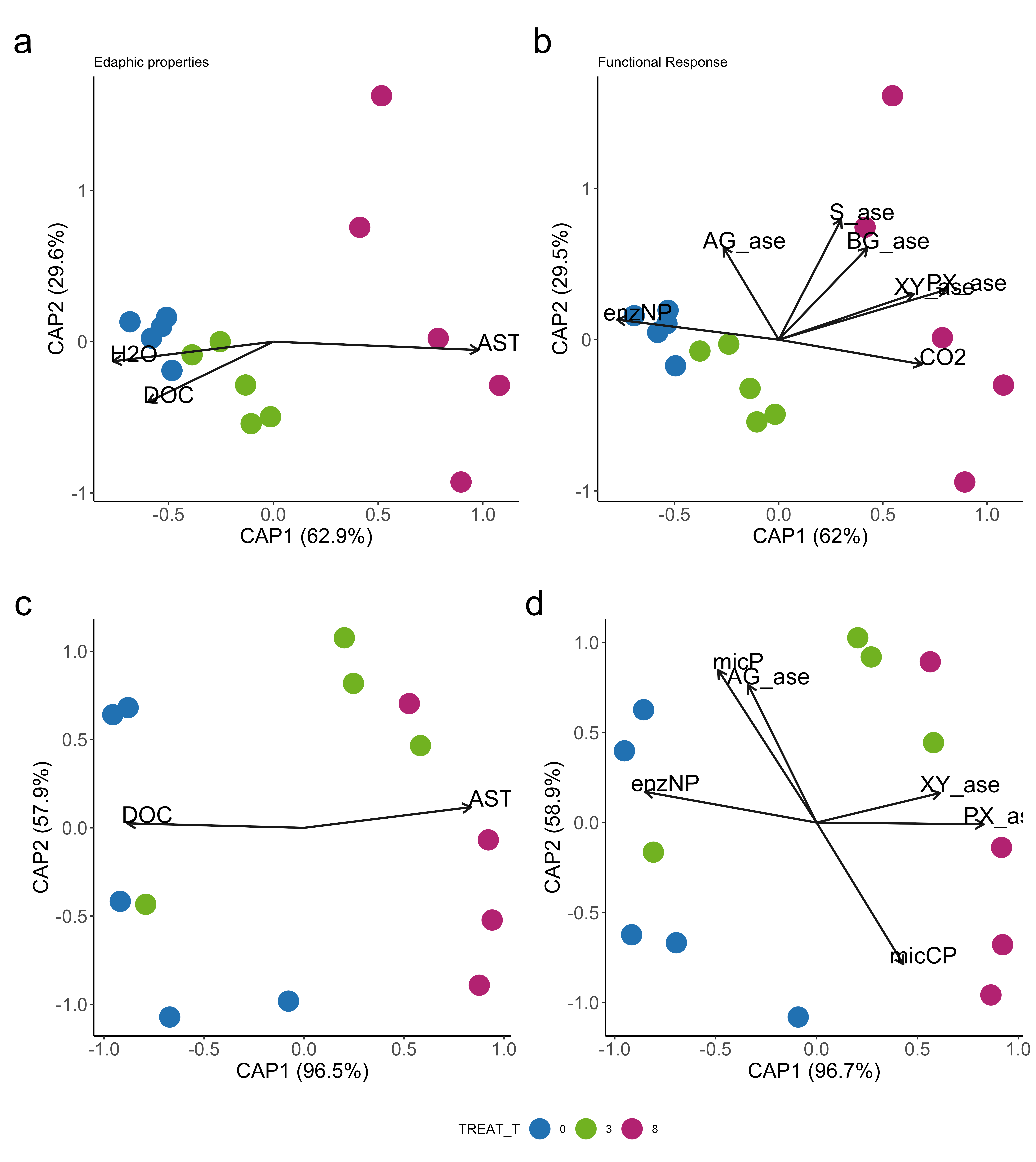
############################################################################# 16S rRNA BETA DIV PLOTS ##################################################################################### First the code for ordination implementation in `phyloseq`. set.seed(119)ssu18_data_sets<-c("ssu18_ps_perfect")ssu_dist<-c("unifrac", "wunifrac")swel_col<-c("#2271B2", "#71B222", "#B22271")for(samp_psinssu18_data_sets){for(dinssu_dist){tmp_get<-get(purrr::map_chr(samp_ps, ~paste0(., "_prop")))ord_meths<-c("PCoA")# MDS = PCoA, "CCA", "DCA", "DPCoA", "RDA"tmp_plist<-plyr::llply(as.list(ord_meths), function(i, physeq, d){ordi=ordinate(physeq, method =i, distance =d)plot_ordination(physeq, ordi, "samples", color ="TEMP")}, tmp_get, d)names(tmp_plist)<-ord_methstmp_df<-plyr::ldply(tmp_plist, function(x){df=x$data[, 1:2]colnames(df)=c("Axis_1", "Axis_2")return(cbind(df, x$data))})names(tmp_df)[1]="method"tmp_plot<-ggplot(tmp_df, aes(Axis_1, Axis_2, color =TEMP, fill =TEMP))tmp_plot<-tmp_plot+geom_point(size =4)tmp_plot<-tmp_plot+facet_wrap(~method, scales ="free")tmp_plot<-tmp_plot+scale_colour_manual(values =swel_col)tmp_df_name<-purrr::map_chr(d, ~paste0(samp_ps, "_dist_", .))tmp_plist_name<-purrr::map_chr(d, ~paste0(samp_ps, "_", ., "_plist"))tmp_plot_name<-purrr::map_chr(d, ~paste0(samp_ps, "_dist_", ., "_plot"))tmp_list<-list("tmp_df_name"=tmp_df, tmp_plist_name =tmp_plist, tmp_plot_name =tmp_plot)assign(paste0(samp_ps, "_", d, "_ord_results"), tmp_list)rm(list =ls(pattern ="_tmp"))}}plist_name<-objects(pattern ="_ord_results")#plot_num <- c(1,2,3,4)plot_num<-c(1)for(iinplist_name){for(jinplot_num){tmp_get_i<-get(i)$tmp_plist_nametmp_ord<-names(tmp_get_i)[j]tmp_name<-stringr::str_replace(i, "ord_results", tmp_ord)tmp_dist<-stringr::str_remove(tmp_name, "ssu18_ps_perfect_")%>%stringr::str_remove(., tmp_ord)%>%stringr::str_remove(., "_")tmp_plot<-tmp_get_i[[j]]+scale_colour_manual(values =swel_col)tmp_plot<-tmp_plot+geom_point(size =4)+theme(legend.position ="none", panel.grid.major =element_blank(), panel.grid.minor =element_blank(), panel.background =element_blank(), axis.line =element_line(colour ="black"))tmp_plot$labels$shape<-"TEMP"if(tmp_dist=="unifrac"){tmp_dist_name<-"Unweighted Unifrac"}elseif(tmp_dist=="wunifrac"){tmp_dist_name<-"Weighted Unifrac"}else{tmp_dist_name<-""}tmp_plot<-tmp_plot+ggtitle(tmp_dist_name)assign(tmp_name, tmp_plot)rm(list =ls(pattern ="tmp_"))}}## And now the code for ordination implementation in `microeco`. rm(self)plot_group_distance_jjs<-function(choose_data, plot_group_order=NULL, color_values=RColorBrewer::brewer.pal(8, "Dark2"), distance_pair_stat=FALSE, hide_ns=FALSE, hide_ns_more=NULL, pair_compare_filter_match=NULL, pair_compare_filter_select=NULL, pair_compare_method="wilcox.test", plot_distance_xtype=NULL){self<-choose_datagroup_distance<-self$res_group_distancegroup<-self$groupif(self$measure%in%c("wei_unifrac", "unwei_unifrac", "bray", "jaccard")){titlename<-switch(self$measure, wei_unifrac ="Weighted Unifrac", unwei_unifrac ="Unweighted Unifrac", bray ="Bray-Curtis", jaccard ="Jaccard")ylabname<-paste0(titlename, " distance")}else{ylabname<-self$measure}if(!is.null(plot_group_order)){group_distance[, group]%<>%factor(., levels =plot_group_order)}else{group_distance[, group]%<>%as.factor}message("The ordered groups are ", paste0(levels(group_distance[, group]), collapse =" "), " ...")p<-ggplot(group_distance, aes_string(x =group, y ="value", color =group))+theme_bw()+theme(panel.grid =element_blank())+geom_boxplot(outlier.size =1, width =0.6, linetype =1)+stat_summary(fun ="mean", geom ="point", shape =20, size =3, fill ="white")+xlab("")+ylab(ylabname)+theme(axis.text =element_text(size =12))+theme(axis.title =element_text(size =17), legend.position ="none")+scale_color_manual(values =color_values)if(!is.null(plot_distance_xtype)){p<-p+theme(axis.text.x =element_text(angle =plot_distance_xtype, colour ="black", vjust =1, hjust =1, size =10))}if(distance_pair_stat==T){comparisons_list<-levels(group_distance[, group])%>%combn(., 2)if(hide_ns){pre_filter<-ggpubr::compare_means(reformulate(group, "value"), group_distance)if(is.null(hide_ns_more)){filter_mark<-"ns"}else{filter_mark<-hide_ns_more}comparisons_list%<>%.[, !(pre_filter$p.signif%in%filter_mark), drop =FALSE]}else{if(!is.null(pair_compare_filter_match)&!is.null(pair_compare_filter_select)){stop("The parameter pair_compare_filter_select and pair_compare_filter_match can not be both used together!")}if(!is.null(pair_compare_filter_match)){comparisons_list%<>%{.[, unlist(lapply(as.data.frame(.), function(x)any(grepl(pair_compare_filter_match, x)))), drop =FALSE]}}if(!is.null(pair_compare_filter_select)){if(!is.numeric(pair_compare_filter_select)){stop("The parameter pair_compare_filter_select must be numeric !")}messages_use<-unlist(lapply(as.data.frame(comparisons_list[, pair_compare_filter_select, drop =FALSE]), function(x){paste0(x, collapse ="-")}))message("Selected groups are ", paste0(messages_use, collapse =" "), " ...")comparisons_list%<>%.[, pair_compare_filter_select, drop =FALSE]}}comparisons_list%<>%{lapply(seq_len(ncol(.)), function(x).[, x])}p<-p+ggpubr::stat_compare_means(comparisons =my_comparisons)}p}my_comparisons<-list(c("0", "3"), c("0", "8"), c("3", "8"))microeco_path<-"include/pub/EXD/"for(iinssu18_data_sets){tmp_dataset<-get(purrr::map_chr(i, ~paste0(., "_me")))tmp_dataset$cal_betadiv(unifrac =TRUE)rm(list =ls(pattern ="tmp_"))}## Here I made a custom "function" to run the analysis, plot the graphs, ## save graph objects, and save plots (as `.png` and `.pdf` files). ## I am sure an actual programmer would be shocked, but it works. microeco_beta_plot<-function(choose_input, choose_metric, choose_ord){tmp_dataset<-get(purrr::map_chr(choose_input, ~paste0(., "_me")))tmp_t1<-trans_beta$new(dataset =tmp_dataset, group ="TEMP", measure =choose_metric)tmp_t1$cal_ordination(ordination =choose_ord)tmp_t1_ord_plot<-tmp_t1$plot_ordination(plot_color ="TEMP", plot_shape ="TEMP", color_values =swel_col, shape_values =c(16, 16, 16))+geom_point(size =4)+theme(legend.position ="none", panel.grid.major =element_blank(), panel.grid.minor =element_blank(), panel.background =element_blank(), axis.line =element_line(colour ="black"))if(choose_metric=="unwei_unifrac"){tmp_plt_name<-"Unweighted Unifrac"}elseif(choose_metric=="wei_unifrac"){tmp_plt_name<-"Weighted Unifrac"}else{tmp_plt_name<-""}tmp_t1_ord_plot<-tmp_t1_ord_plot+ggtitle(tmp_plt_name)tmp_t1$cal_group_distance()tmp_t1$plot_group_distance_jjs<-plot_group_distance_jjstmp_t1_within_group_plot<-tmp_t1$plot_group_distance_jjs(choose_data =tmp_t1, distance_pair_stat =TRUE, color_values =swel_col)tmp_t1_within_group_plot<-tmp_t1_within_group_plot+ggtitle(tmp_plt_name)tmp_t1$res_group_distancetmp_t1$cal_group_distance(within_group =FALSE)tmp_t1_btwn_group_plot<-tmp_t1$plot_group_distance_jjs(choose_data =tmp_t1, distance_pair_stat =TRUE, color_values =swel_col)tmp_t1_btwn_group_plot<-tmp_t1_btwn_group_plot+ggtitle(tmp_plt_name)###### SET namestmp_name_ord<-paste(choose_input, "_me_", choose_metric, "_", choose_ord, sep ="")tmp_name_wg<-paste(choose_input, "_me_wg_", choose_metric, "_", choose_ord, sep ="")tmp_name_bg<-paste(choose_input, "_me_bg_", choose_metric, "_", choose_ord, sep ="")assign(tmp_name_ord, tmp_t1_ord_plot, envir =parent.frame())assign(tmp_name_wg, tmp_t1_within_group_plot, envir =parent.frame())assign(tmp_name_bg, tmp_t1_btwn_group_plot, envir =parent.frame())rm(list =ls(pattern ="_PCoA"))}for(jin1:length(get(paste("ssu18_ps_perfect", "_me", sep =""))$beta_diversity)){tmp_metric<-names(get(paste("ssu18_ps_perfect", "_me", sep =""))$beta_diversity[j])microeco_beta_plot(choose_input ="ssu18_ps_perfect", choose_metric =tmp_metric, choose_ord ="PCoA")rm(list =ls(pattern ="tmp_"))}ssu18_unifrac<-ssu18_ps_perfect_unifrac_PCoA+geom_point(size =7)ssu18_wunifrac<-ssu18_ps_perfect_wunifrac_PCoA+geom_point(size =7)ssu18_wg_unwei_unifrac<-ssu18_ps_perfect_me_wg_unwei_unifrac_PCoAssu18_wg_wei_unifrac<-ssu18_ps_perfect_me_wg_wei_unifrac_PCoAssu18_wg_unwei_unifrac<-ssu18_wg_unwei_unifrac+theme(axis.title.y =element_text(size =10))+ylab("distance")ssu18_wg_wei_unifrac<-ssu18_wg_wei_unifrac+theme(axis.title.y =element_text(size =10))+ylab("distance")############################################################################# ITS BETA DIV PLOTS #######################################################################################set.seed(119)its18_data_sets<-c("its18_ps_perfect")its_dist<-c("jsd", "bray")swel_col<-c("#2271B2", "#71B222", "#B22271")for(samp_psinits18_data_sets){for(dinits_dist){tmp_get<-get(purrr::map_chr(samp_ps, ~paste0(., "_prop")))ord_meths<-c("PCoA")# MDS = PCoA, "CCA", "DCA", "DPCoA", "RDA"tmp_plist<-plyr::llply(as.list(ord_meths), function(i, physeq, d){ordi=ordinate(physeq, method =i, distance =d)plot_ordination(physeq, ordi, "samples", color ="TEMP")}, tmp_get, d)names(tmp_plist)<-ord_methstmp_df<-plyr::ldply(tmp_plist, function(x){df=x$data[, 1:2]colnames(df)=c("Axis_1", "Axis_2")return(cbind(df, x$data))})names(tmp_df)[1]="method"tmp_plot<-ggplot(tmp_df, aes(Axis_1, Axis_2, color =TEMP, fill =TEMP))tmp_plot<-tmp_plot+geom_point(size =4)tmp_plot<-tmp_plot+facet_wrap(~method, scales ="free")tmp_plot<-tmp_plot+scale_colour_manual(values =swel_col)tmp_df_name<-purrr::map_chr(d, ~paste0(samp_ps, "_dist_", .))tmp_plist_name<-purrr::map_chr(d, ~paste0(samp_ps, "_", ., "_plist"))tmp_plot_name<-purrr::map_chr(d, ~paste0(samp_ps, "_dist_", ., "_plot"))tmp_list<-list("tmp_df_name"=tmp_df, tmp_plist_name =tmp_plist, tmp_plot_name =tmp_plot)assign(paste0(samp_ps, "_", d, "_ord_results"), tmp_list)rm(list =ls(pattern ="_tmp"))}}plist_name<-objects(pattern ="_ord_results")#plot_num <- c(1,2,3,4)plot_num<-c(1)for(iinplist_name){for(jinplot_num){tmp_get_i<-get(i)$tmp_plist_nametmp_ord<-names(tmp_get_i)[j]tmp_name<-stringr::str_replace(i, "ord_results", tmp_ord)tmp_dist<-stringr::str_remove(tmp_name, "its18_ps_perfect_")%>%stringr::str_remove(., tmp_ord)%>%stringr::str_remove(., "_")tmp_plot<-tmp_get_i[[j]]+scale_colour_manual(values =swel_col)tmp_plot<-tmp_plot+geom_point(size =4)+theme(legend.position ="none", panel.grid.major =element_blank(), panel.grid.minor =element_blank(), panel.background =element_blank(), axis.line =element_line(colour ="black"))tmp_plot$labels$shape<-"TEMP"if(tmp_dist=="jsd"){tmp_dist_name<-"Jensen-Shannon"}elseif(tmp_dist=="bray"){tmp_dist_name<-"Bray-Curtis"}else{tmp_dist_name<-""}tmp_plot<-tmp_plot+ggtitle(tmp_dist_name)assign(tmp_name, tmp_plot)rm(list =ls(pattern ="tmp_"))}}## And now the code for ordination implementation in `microeco`. my_comparisons<-list(c("0", "3"), c("0", "8"), c("3", "8"))microeco_path<-"include/pub/EXD/"for(iinits18_data_sets){tmp_dataset<-get(purrr::map_chr(i, ~paste0(., "_me")))tmp_dataset$cal_betadiv(unifrac =FALSE)#### CODE TO ADD JSD DISTANCE #### tmp_jsd<-phyloseq::distance(get(i), method ="jsd")tmp_jsd<-forceSymmetric(as.matrix(tmp_jsd), uplo ="L")tmp_jsd<-as.matrix(tmp_jsd)tmp_dataset$beta_diversity$jsd<-tmp_jsdrm(list =ls(pattern ="tmp_"))}## Here I made a custom "function" to run the analysis, plot the graphs, ## save graph objects, and save plots (as `.png` and `.pdf` files). ## I am sure an actual programmer would be shocked, but it works. microeco_beta_plot<-function(choose_input, choose_metric, choose_ord){tmp_dataset<-get(purrr::map_chr(choose_input, ~paste0(., "_me")))tmp_t1<-trans_beta$new(dataset =tmp_dataset, group ="TEMP", measure =choose_metric)tmp_t1$cal_ordination(ordination =choose_ord)tmp_t1_ord_plot<-tmp_t1$plot_ordination(plot_color ="TEMP", plot_shape ="TEMP", color_values =swel_col, shape_values =c(16, 16, 16))+geom_point(size =4)+theme(legend.position ="none", panel.grid.major =element_blank(), panel.grid.minor =element_blank(), panel.background =element_blank(), axis.line =element_line(colour ="black"))if(choose_metric=="jsd"){tmp_plt_name<-"Jensen-Shannon"}elseif(choose_metric=="bray"){tmp_plt_name<-"Bray-Curtis"}else{tmp_plt_name<-""}tmp_t1_ord_plot<-tmp_t1_ord_plot+ggtitle(tmp_plt_name)tmp_t1$cal_group_distance()tmp_t1$plot_group_distance_jjs<-plot_group_distance_jjstmp_t1_within_group_plot<-tmp_t1$plot_group_distance_jjs(choose_data =tmp_t1, distance_pair_stat =TRUE, color_values =swel_col)tmp_t1_within_group_plot<-tmp_t1_within_group_plot+ggtitle(tmp_plt_name)tmp_t1$res_group_distancetmp_t1$cal_group_distance(within_group =FALSE)tmp_t1_btwn_group_plot<-tmp_t1$plot_group_distance_jjs(choose_data =tmp_t1, distance_pair_stat =TRUE, color_values =swel_col)tmp_t1_btwn_group_plot<-tmp_t1_btwn_group_plot+ggtitle(tmp_plt_name)###### SET namestmp_name_ord<-paste(choose_input, "_me_", choose_metric, "_", choose_ord, sep ="")tmp_name_wg<-paste(choose_input, "_me_wg_", choose_metric, "_", choose_ord, sep ="")tmp_name_bg<-paste(choose_input, "_me_bg_", choose_metric, "_", choose_ord, sep ="")assign(tmp_name_ord, tmp_t1_ord_plot, envir =parent.frame())assign(tmp_name_wg, tmp_t1_within_group_plot, envir =parent.frame())assign(tmp_name_bg, tmp_t1_btwn_group_plot, envir =parent.frame())rm(list =ls(pattern ="_PCoA"))}for(jin1:length(get(paste("its18_ps_perfect", "_me", sep =""))$beta_diversity)){tmp_metric<-names(get(paste("its18_ps_perfect", "_me", sep =""))$beta_diversity[j])microeco_beta_plot(choose_input ="its18_ps_perfect", choose_metric =tmp_metric, choose_ord ="PCoA")rm(list =ls(pattern ="tmp_"))}its18_jsd<-its18_ps_perfect_jsd_PCoA+geom_point(size =7)its18_bray<-its18_ps_perfect_bray_PCoA+geom_point(size =7)its18_wg_jsd<-its18_ps_perfect_me_wg_jsd_PCoAits18_wg_bray<-its18_ps_perfect_me_wg_bray_PCoAits18_wg_jsd<-its18_wg_jsd+theme(axis.title.y =element_text(size =10))+ylab("distance")its18_wg_bray<-its18_wg_bray+theme(axis.title.y =element_text(size =10))+ylab("distance")### CREATE combo plot using PATCHWORKlayout<-c(area(t =1, b =15, l =1, r =22),area(t =17, b =31, l =2, r =9),area(t =17, b =31, l =10, r =17),area(t =16, b =23, l =18, r =21),area(t =24, b =31, l =18, r =21),area(t =32, b =46, l =1, r =22),area(t =48, b =62, l =2, r =9),area(t =48, b =62, l =10, r =17),area(t =47, b =54, l =18, r =21),area(t =55, b =62, l =18, r =21))plot(layout)combo_plot<-ssu18_alph_div_plots_asv+ssu18_unifrac+ssu18_wunifrac+ssu18_wg_unwei_unifrac+ssu18_wg_wei_unifrac+its18_alph_div_plots_asv+its18_jsd+its18_bray+its18_wg_jsd+its18_wg_bray+plot_layout(design =layout)ggplot2::ggsave(combo_plot, path ="include/pub/EXD/EXD_Figure_2/", filename ="Extended_Data_Figure_2.png", height =56, width =40, units ='cm', dpi =600, bg ="white")ggplot2::ggsave(combo_plot, path ="include/pub/EXD/EXD_Figure_2/", filename ="Extended_Data_Figure_2.pdf", height =56, width =40, units ='cm', dpi =600, bg ="white")
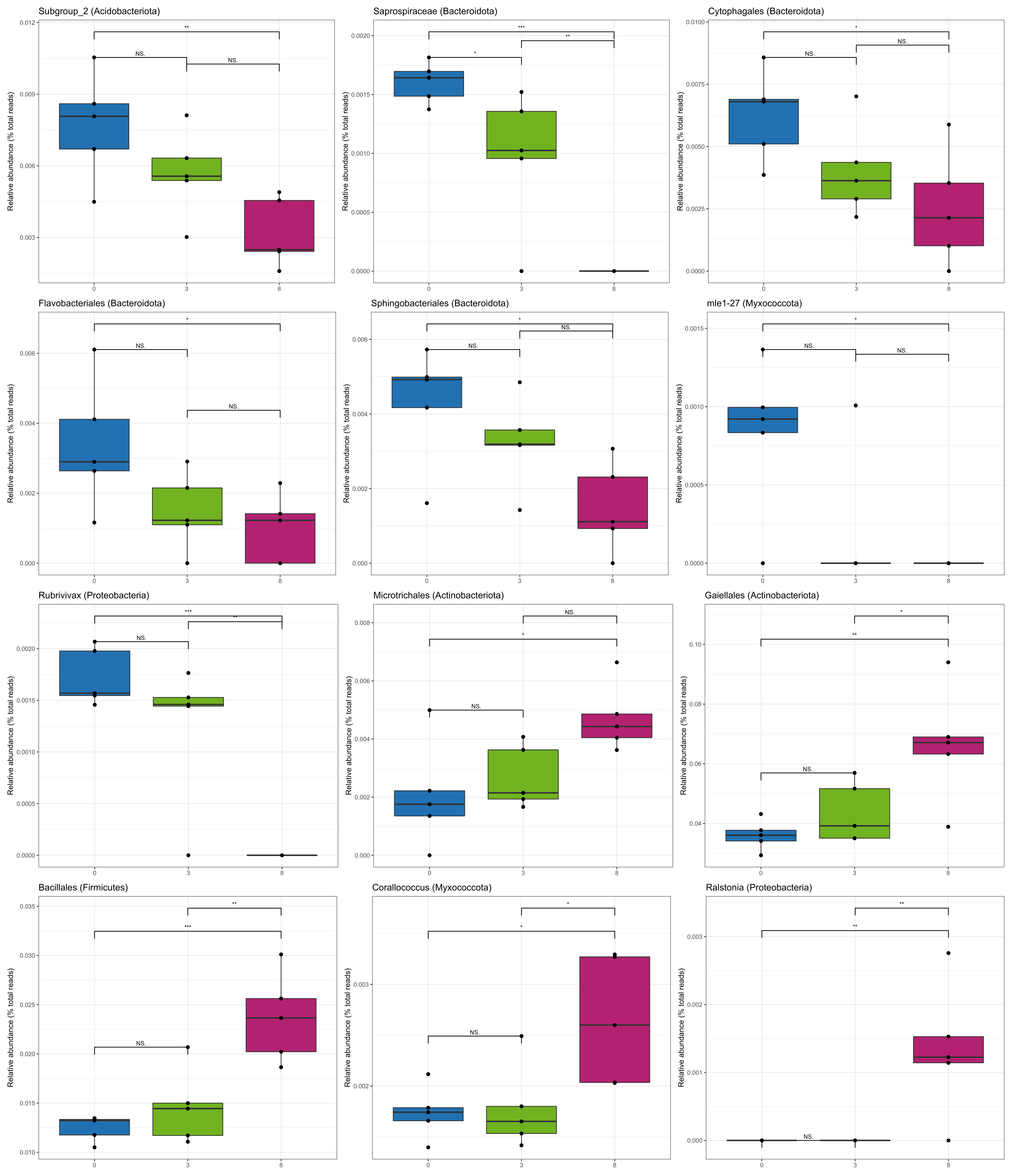
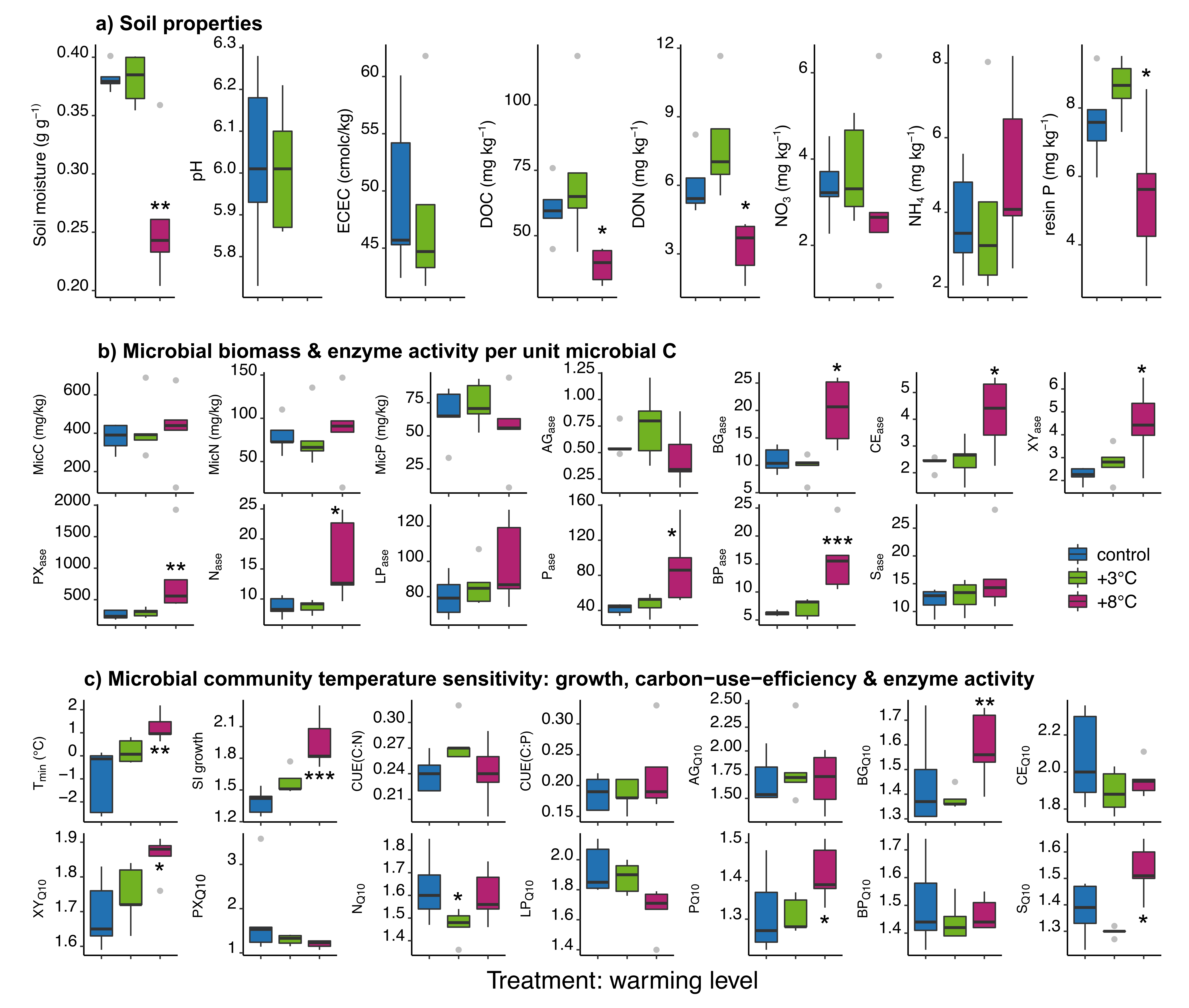
Extended Data Figure 3
Modifications
Post processing performed in Inkscape. Modifications include axes renaming, removing non-significant (NS) values from plots, changing significant p-values to asterisks (*), removing gridlines, and enlarging outlier points.
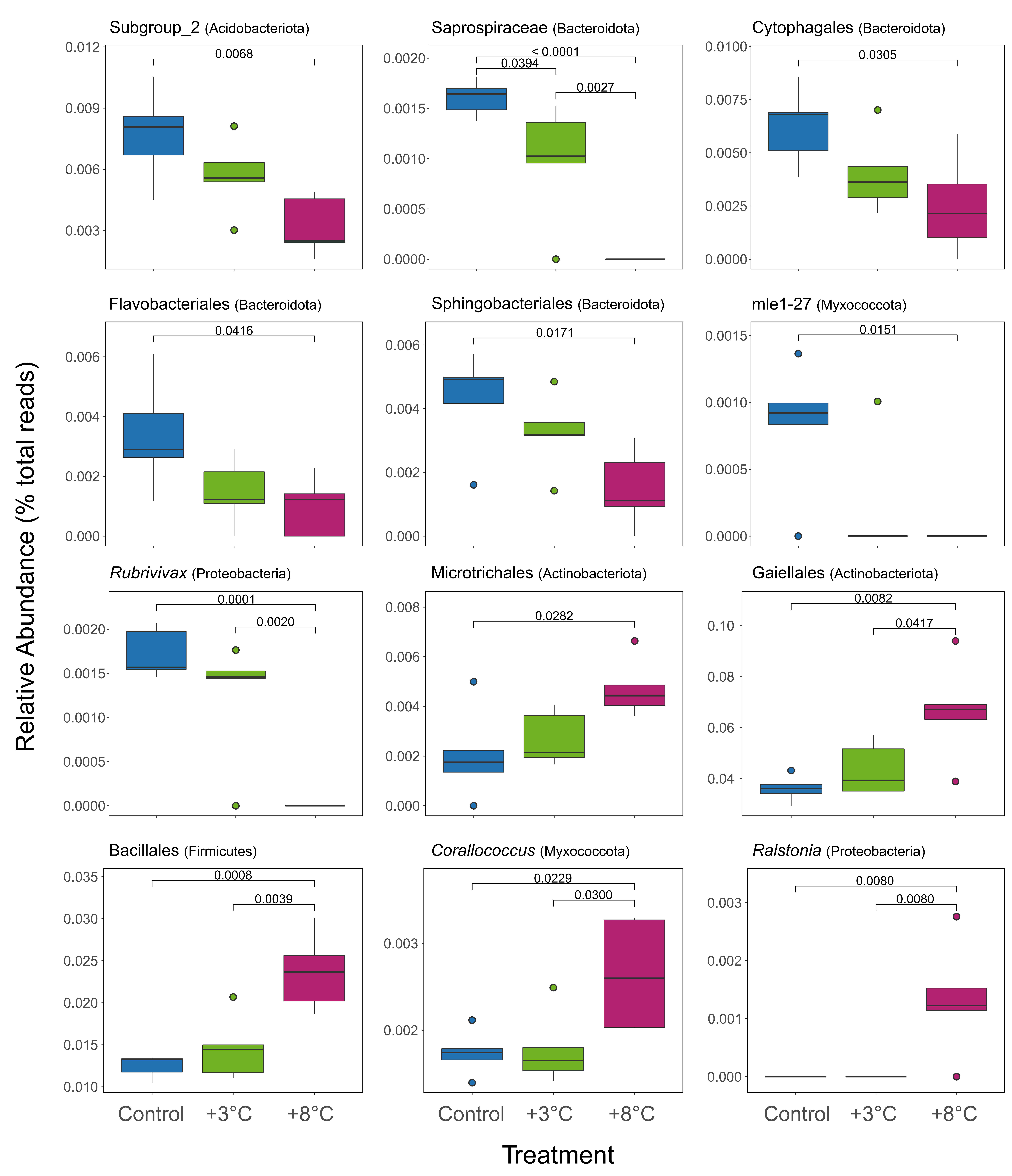
## In this section of the workflow we use the ## [`microbiomeMarker`](https://github.com/yiluheihei/microbiomeMarker) package ## to assess the response of taxonomic lineages to soil warming. ## In the first step we need to fix the selected data set to make it ## compatible with the various functions. For this analysis we use the ## PERfect filtered data set.remove(list =ls())ssu18_ps_perfect_rf_all<-readRDS("include/pub/EXD/EXD_Figure_3/Extended_Data_Figure_3.rds")## FIX ps objectssu_ps<-ssu18_ps_perfect_rf_alltmp_tax1<-data.frame(tax_table(ssu_ps))tmp_rn<-row.names(tmp_tax1)tmp_tax<-data.frame(lapply(tmp_tax1, function(x){gsub("\\(|)", "", x)}))row.names(tmp_tax)<-tmp_rnidentical(row.names(tmp_tax), row.names(tmp_tax1))ps_tax_new<-as.matrix(tmp_tax)tmp_ps<-phyloseq(otu_table(ssu_ps),phy_tree(ssu_ps),tax_table(ps_tax_new),sample_data(ssu_ps))ssu_ps<-tmp_psphyloseq::rank_names(ssu_ps)## Next we run a statistical test for multiple groups## using the `run_test_multiple_groups` function.ssu_group_anova<-run_test_multiple_groups(ssu_ps, group ="TEMP", taxa_rank ="all", method ="anova")ssu_group_anova@marker_tablemarker_table(ssu_group_anova)## And then conduct post hoc pairwise comparisons for multiple## groups test using the `run_posthoc_test` function.ssu_default_pht<-run_posthoc_test(ssu_ps, group ="TEMP", method ="tukey", transform ="log10")## We can filter out a select taxa and plot the results.filter(data.frame(ssu_default_pht@result),group_name=="k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales")plot_postHocTest(ssu_default_pht, feature ="k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales")&theme_bw()## But what we really want to do is get all of the markers that are## significant from the analysis, excluding any significant ASVs so we can## look at high taxa ranks.ssu_pht<-ssu_default_phtssu18_pht_filt<-filter(data.frame(ssu_pht@result), pvalue<="0.05")[!grepl("ASV", filter(data.frame(ssu_pht@result), pvalue<="0.05")$group_name), ]ssu18_pht_filt<-ssu18_pht_filt[!grepl("[a-z]__$", ssu18_pht_filt$group_name), ]ssu18_pht_filt<-distinct(ssu18_pht_filt, group_name, .keep_all =TRUE)nrow(ssu18_pht_filt)plot_postHocTest_jjs<-function(pht, feature, step_increase=0.12){abd_long<-pht@abundance%>%tidyr::pivot_longer(-.data$group, names_to ="feat")if(!is.null(feature)){abd_long<-filter(abd_long, .data$feat%in%feature)}annotation<-get_sig_annotation(pht, step_increase =step_increase)p_box<-suppressWarnings(ggplot(abd_long, aes(x =.data$group, y =.data$value))+geom_boxplot()+ggsignif::geom_signif(data =annotation[annotation$feature%in%feature, ], aes(xmin =.data$xmin, xmax =.data$xmax, annotations =.data$annotation, y_position =.data$y_position), manual =TRUE, textsize =3, vjust =0.2)+labs(x =NULL, y ="Abundance"))test_res<-as.data.frame(pht@result[[feature]])p_test<-ggplot(test_res, aes(x =.data$comparions))+geom_errorbar(aes(ymin =.data$ci_lower, ymax =.data$ci_upper), width =0.2)+geom_point(aes(y =.data$diff_mean))+labs(x =NULL, y ="95% confidence intervals")patchwork::wrap_plots(p_box)}environment(plot_postHocTest_jjs)<-asNamespace('microbiomeMarker')ssu_select<-c("k__Bacteria|p__Acidobacteriota|c__Acidobacteriae|o__Subgroup_2", "k__Bacteria|p__Bacteroidota|c__Bacteroidia|o__Chitinophagales|f__Saprospiraceae", "k__Bacteria|p__Bacteroidota|c__Bacteroidia|o__Cytophagales", "k__Bacteria|p__Bacteroidota|c__Bacteroidia|o__Flavobacteriales", "k__Bacteria|p__Bacteroidota|c__Bacteroidia|o__Sphingobacteriales", "k__Bacteria|p__Myxococcota|c__Polyangia|o__mle1-27", "k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Burkholderiales|f__Comamonadaceae|g__Rubrivivax", "k__Bacteria|p__Actinobacteriota|c__Acidimicrobiia|o__Microtrichales", "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales", "k__Bacteria|p__Firmicutes|c__Bacilli|o__Bacillales", "k__Bacteria|p__Myxococcota|c__Myxococcia|o__Myxococcales|f__Myxococcaceae|g__Corallococcus", "k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Burkholderiales|f__Burkholderiaceae|g__Ralstonia")swel_col<-c("#2271B2", "#71B222", "#B22271")for(iinssu_select){tmp_select_feat<-itmp_plot<-plot_postHocTest_jjs(ssu_pht, feature =tmp_select_feat)&theme_bw()tmp_plot<-tmp_plot+geom_boxplot(fill =swel_col)+scale_colour_manual(values =c("#191919", "#191919", "#191919"))+geom_point(size =2, show.legend =FALSE)+ylab("Relative abundance (% total reads)")tmp_name<-purrr::map_chr(i, ~paste0(., "_tax_plot"))assign(tmp_name, tmp_plot)rm(list =ls(pattern ="tmp_"))}ssu_title<-c("Subgroup_2 (Acidobacteriota)", "Saprospiraceae (Bacteroidota)", "Cytophagales (Bacteroidota)", "Flavobacteriales (Bacteroidota)", "Sphingobacteriales (Bacteroidota)", "mle1-27 (Myxococcota)", "Rubrivivax (Proteobacteria)", "Microtrichales (Actinobacteriota)", "Gaiellales (Actinobacteriota)", "Bacillales (Firmicutes)", "Corallococcus (Myxococcota)", "Ralstonia (Proteobacteria)")ssu_plt_info<-data.frame(lineage =ssu_select, label =ssu_title)for(iinseq_len(nrow(ssu_plt_info))){tmp_name<-paste("plot_", i, sep ="")tmp_plot<-get(purrr::map_chr(ssu_plt_info[i, 1], ~paste0(., "_tax_plot")))+geom_point(show.legend =FALSE)+ggtitle(ssu_plt_info[i, 2])assign(tmp_name, tmp_plot)rm(list =ls(pattern ="tmp_"))}ssu_taxa_combo_plot<-((plot_1+plot_2+plot_3)/(plot_4+plot_5+plot_6)/(plot_7+plot_8+plot_9)/(plot_10+plot_11+plot_12))ggplot2::ggsave(ssu_taxa_combo_plot, path ="include/pub/EXD/EXD_Figure_3/", filename ="Extended_Data_Figure_3.png", height =14157, width =12186, units ='px', bg ="white", dpi =600)ggplot2::ggsave(ssu_taxa_combo_plot, path ="include/pub/EXD/EXD_Figure_3/", filename ="Extended_Data_Figure_3.pdf", height =14157, width =12186, units ='px', bg ="white", dpi =600)
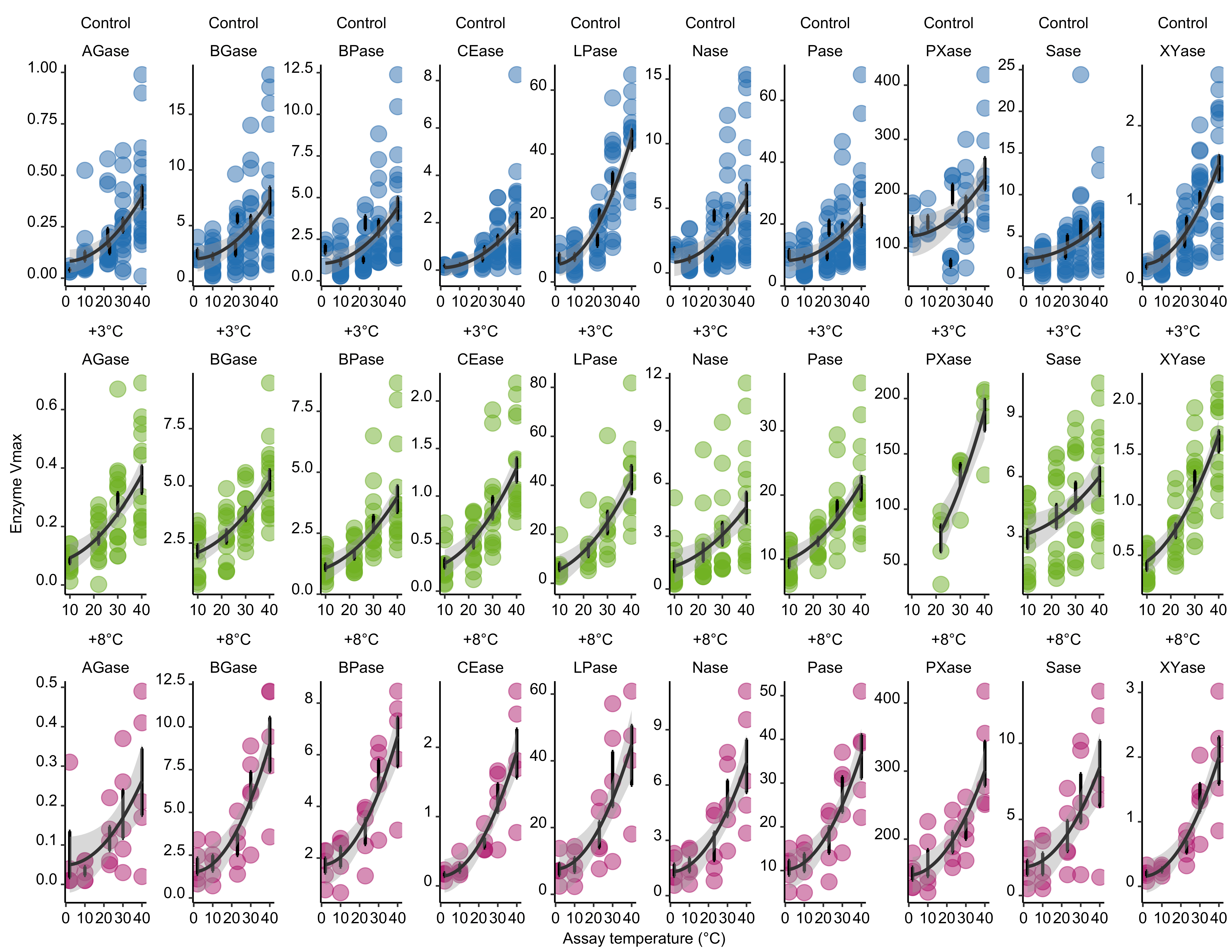
Extended Data Figure 4
Modifications
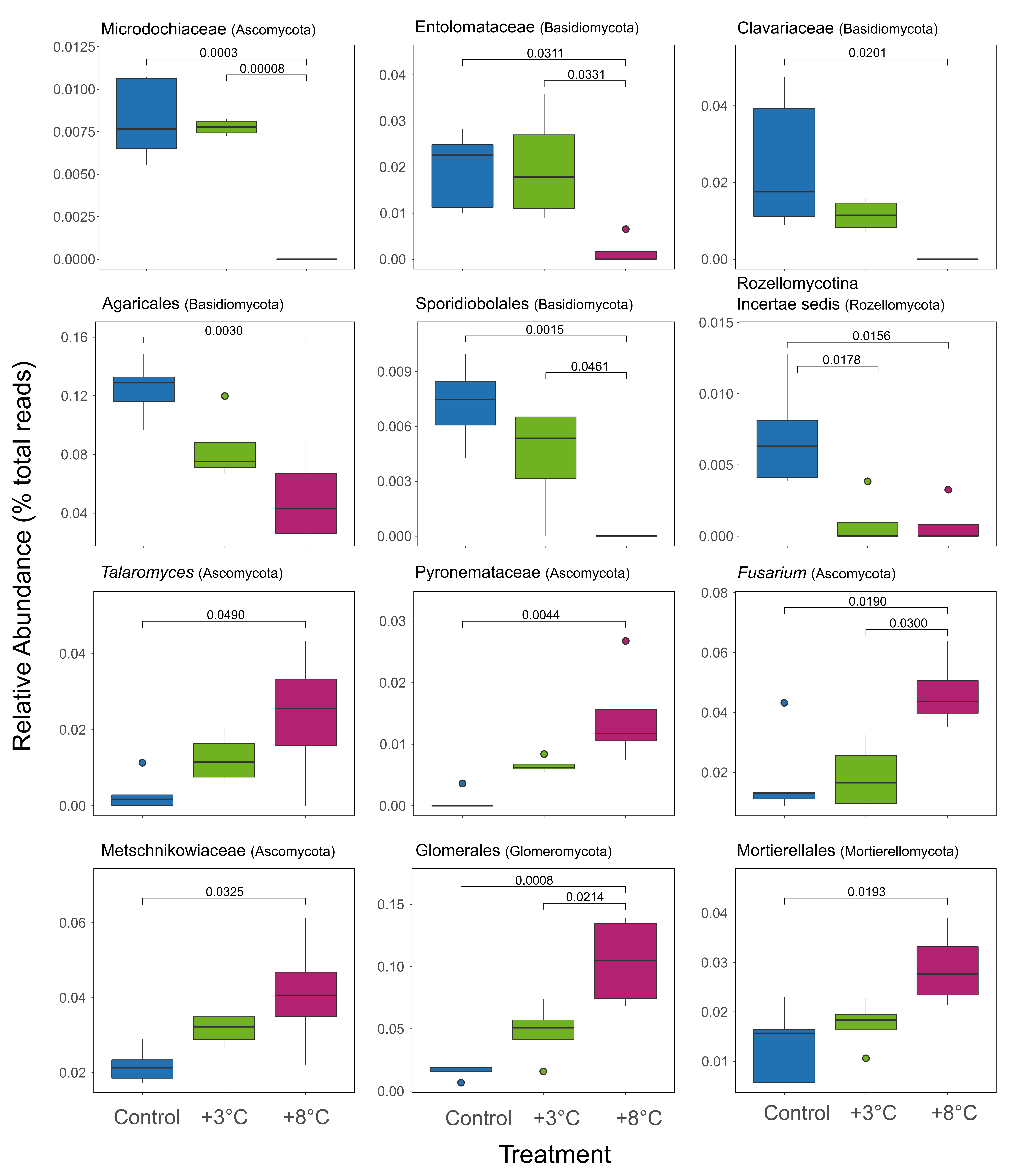
Post processing performed in Inkscape. Modifications include axes renaming, removing non-significant (NS) values from plots, changing significant p-values to asterisks (*), removing gridlines, and enlarging outlier points.
## In this section of the workflow we use the ## [`microbiomeMarker`](https://github.com/yiluheihei/microbiomeMarker) package ## to assess the response of taxonomic lineages to soil warming. ## In the first step we need to fix the selected data set to make it ## compatible with the various functions. For this analysis we use the ## PERfect filtered data set.remove(list =ls())its18_ps_perfect_rf_all<-readRDS("include/pub/EXD/EXD_Figure_4/Extended_Data_Figure_4.rds")## FIX ps objectits_ps<-its18_ps_perfect_rf_alltmp_tax1<-data.frame(tax_table(its_ps))tmp_tax1$ASV_SEQ<-NULLtmp_rn<-row.names(tmp_tax1)tmp_tax<-data.frame(lapply(tmp_tax1, function(x){gsub("\\(|)", "", x)}))row.names(tmp_tax)<-tmp_rnidentical(row.names(tmp_tax), row.names(tmp_tax1))ps_tax_new<-as.matrix(tmp_tax)tmp_ps<-phyloseq(otu_table(its_ps),tax_table(ps_tax_new),sample_data(its_ps))its_ps<-tmp_psphyloseq::rank_names(its_ps)## Next we run a statistical test for multiple groups## using the `run_test_multiple_groups` function.its_group_anova<-run_test_multiple_groups(its_ps, group ="TEMP", taxa_rank ="all", method ="anova")its_group_anova@marker_tablemarker_table(its_group_anova)## And then conduct post hoc pairwise comparisons for multiple## groups test using the `run_posthoc_test` function.its_default_pht<-run_posthoc_test(its_ps, group ="TEMP", method ="tukey", transform ="log10")## But what we really want to do is get all of the markers that are## significant from the analysis, excluding any significant ASVs so we can## look at high taxa ranks.its_pht<-its_default_phtits18_pht_filt<-filter(data.frame(its_pht@result), pvalue<="0.05")[!grepl("ASV", filter(data.frame(its_pht@result), pvalue<="0.05")$group_name),]its18_pht_filt<-its18_pht_filt[!grepl("[a-z]__$", its18_pht_filt$group_name),]its18_pht_filt<-unique(its18_pht_filt$group_name)plot_postHocTest_jjs<-function(pht, feature, step_increase=0.12){abd_long<-pht@abundance%>%tidyr::pivot_longer(-.data$group, names_to ="feat")if(!is.null(feature)){abd_long<-filter(abd_long, .data$feat%in%feature)}annotation<-get_sig_annotation(pht, step_increase =step_increase)p_box<-suppressWarnings(ggplot(abd_long, aes(x =.data$group, y =.data$value))+geom_boxplot()+ggsignif::geom_signif(data =annotation[annotation$feature%in%feature, ], aes(xmin =.data$xmin, xmax =.data$xmax, annotations =.data$annotation, y_position =.data$y_position), manual =TRUE, textsize =3, vjust =0.2)+labs(x =NULL, y ="Abundance"))test_res<-as.data.frame(pht@result[[feature]])p_test<-ggplot(test_res, aes(x =.data$comparions))+geom_errorbar(aes(ymin =.data$ci_lower, ymax =.data$ci_upper), width =0.2)+geom_point(aes(y =.data$diff_mean))+labs(x =NULL, y ="95% confidence intervals")patchwork::wrap_plots(p_box)}environment(plot_postHocTest_jjs)<-asNamespace('microbiomeMarker')its_select<-c("k__Fungi|p__Ascomycota|c__Sordariomycetes|o__Xylariales|f__Microdochiaceae", "k__Fungi|p__Basidiomycota|c__Agaricomycetes|o__Agaricales|f__Entolomataceae", "k__Fungi|p__Basidiomycota|c__Agaricomycetes|o__Agaricales|f__Clavariaceae", "k__Fungi|p__Basidiomycota|c__Agaricomycetes|o__Agaricales", "k__Fungi|p__Basidiomycota|c__Microbotryomycetes|o__Sporidiobolales", "k__Fungi|p__Rozellomycota|c__Rozellomycotina_cls_Incertae_sedis", "k__Fungi|p__Ascomycota|c__Eurotiomycetes|o__Eurotiales|f__Trichocomaceae|g__Talaromyces", "k__Fungi|p__Ascomycota|c__Pezizomycetes|o__Pezizales|f__Pyronemataceae", "k__Fungi|p__Ascomycota|c__Sordariomycetes|o__Hypocreales|f__Nectriaceae|g__Fusarium", "k__Fungi|p__Ascomycota|c__Saccharomycetes|o__Saccharomycetales|f__Metschnikowiaceae", "k__Fungi|p__Glomeromycota|c__Glomeromycetes|o__Glomerales", "k__Fungi|p__Mortierellomycota|c__Mortierellomycetes|o__Mortierellales")swel_col<-c("#2271B2", "#71B222", "#B22271")for(iinits_select){tmp_select_feat<-itmp_plot<-plot_postHocTest_jjs(its_pht, feature =tmp_select_feat)&theme_bw()tmp_plot<-tmp_plot+geom_boxplot(fill =swel_col)+scale_colour_manual(values =c("#191919", "#191919", "#191919"))+geom_point(size =2, show.legend =FALSE)+ylab("Relative abundance (% total reads)")tmp_name<-purrr::map_chr(i, ~paste0(., "_tax_plot"))assign(tmp_name, tmp_plot)rm(list =ls(pattern ="tmp_"))}its_title<-c("Microdochiaceae (Ascomycota)", "Entolomataceae (Basidiomycota)", "Clavariaceae (Basidiomycota)", "Agaricales (Basidiomycota)", "Sporidiobolales (Basidiomycota)", "Rozellomycotina (Rozellomycota)", "Talaromyces (Ascomycota)", "Pyronemataceae (Ascomycota)", "Fusarium (Ascomycota)", "Metschnikowiaceae (Ascomycota)", "Glomerales (Glomeromycota)", "Mortierellales (Mortierellomycota)")its_plt_info<-data.frame(lineage =its_select, label =its_title)for(iinseq_len(nrow(its_plt_info))){tmp_name<-paste("plot_", i, sep ="")tmp_plot<-get(purrr::map_chr(its_plt_info[i, 1], ~paste0(., "_tax_plot")))+geom_point(show.legend =FALSE)+ggtitle(its_plt_info[i, 2])assign(tmp_name, tmp_plot)rm(list =ls(pattern ="tmp_"))}its_taxa_combo_plot<-((plot_1+plot_2+plot_3)/(plot_4+plot_5+plot_6)/(plot_7+plot_8+plot_9)/(plot_10+plot_11+plot_12))ggplot2::ggsave(its_taxa_combo_plot, path ="include/pub/EXD/EXD_Figure_4/", filename ="Extended_Data_Figure_4.png", height =14157, width =12186, units ='px', bg ="white", dpi =600)ggplot2::ggsave(its_taxa_combo_plot, path ="include/pub/EXD/EXD_Figure_4/", filename ="Extended_Data_Figure_4.pdf", height =14157, width =12186, units ='px', bg ="white", dpi =600)
Extended Data Figure 5
Modifications
Post processing performed in Inkscape. Modifications include repositioning vector overlay labels, styling legend, and changing font size and style.
remove(list =ls())load("include/pub/EXD/EXD_Figure_5/Extended_Data_Figure_5.rdata")## 1) Run `rankindex` to compare metadata and community dissimilarity indices ## for gradient detection. This will help us select the best dissimilarity ## metric to use.## 2) Run `capscale` for distance-based redundancy analysis.## 3) Run `envfit` to fit environmental parameters onto the ordination. ## This function basically calculates correlation scores between the metadata ## parameters and the ordination axes. ## 4) Select metadata parameters significant for `bioenv` (see above) ## and/or `envfit` analyses.## 5) Run `envfit` on ASVs.## 6) Plot the ordination and vector overlays. ############################################################################# 16S rRNA EDAPHIC PROPERTIES ###############################################################################tmp_md<-ssu18_select_mc_norm_split_no_ac$edaphictmp_md$TREAT_T<-as.character(tmp_md$TREAT_T)tmp_comm<-data.frame(t(ssu18_select_mc_norm_split_no_ac$data_loaded))edaphic_rank<-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow","bra", "kul"), stepacross =FALSE, method ="spearman")## Run `capscale` using Bray-Curtis. ## * Starting properties: AST, H2O, N, P, Al, Ca, Fe, K, Mg, Mn, Na, TEB, ECEC, pH, NH4, NO3, PO4, DOC, DON, DOCN## * Autocorrelated removed: TEB, DON, Na, Al, Ca## 15 total, only works with 13## * Remove for capscale: Mg, Mnedaphic_cap<-capscale(tmp_comm~AST+H2O+N+P+Fe+K+ECEC+pH+NH4+NO3+PO4+DOC+DOCN, tmp_md, dist ="bray")anova(edaphic_cap)# overall test of the significant of the analysisanova(edaphic_cap, by ="axis", perm.max =500)# test axes for significanceanova(edaphic_cap, by ="terms", permu =500)# test for sign. environ. variables## Next, we need to grab capscale scores for the samples and create a ## data frame of the first two dimensions. We will also need to add some ## of the sample details to the data frame. For this we use the vegan ## function `scores` which gets species or site scores from the ordination.library(ggvegan)tmp_auto_plt<-ggplot2::autoplot(edaphic_cap, arrows =TRUE)tmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")edaphic_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")## Now we have a new data frame that contains sample details and capscale values. ## We can then do the same with the metadata vectors. ## Here though we only need the scores and parameter name. edaphic_md_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="biplot")edaphic_md_scores[,1]<-NULLedaphic_md_scores<-edaphic_md_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")## Let's run some quick correlations of metadata with ordination axes to ## see which parameters are significant. For this we use the vegan function `envfit`.tmp_samp_scores_sub<-edaphic_plot_data[, 6:7]tmp_samp_scores_sub<-as.matrix(tmp_samp_scores_sub)tmp_param_list<-edaphic_md_scores$parameterstmp_md_sub<-subset(tmp_md, select =tmp_param_list)envfit_edaphic_md<-envfit(tmp_samp_scores_sub, tmp_md_sub, perm =1000, choices =c(1, 2))edaphic_md_signif_hits<-base::subset(envfit_edaphic_md$vectors$pvals, c(envfit_edaphic_md$vectors$pvals<0.05&envfit_edaphic_md$vectors$r>0.4))edaphic_md_signif_hits<-data.frame(edaphic_md_signif_hits)edaphic_md_signif_hits<-rownames(edaphic_md_signif_hits)edaphic_md_signif<-edaphic_md_scores[edaphic_md_scores$parameters%in%edaphic_md_signif_hits,]## Now let's see if the same parameters are significant for the `envfit` and `bioenv` analyses.print("Significant parameters from bioenv analysis.")row.names(summary(ssu18_edaphic_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")edaphic_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(ssu18_edaphic_bioenv_ind_mantel)), edaphic_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(edaphic_md_signif$parameters, row.names(summary(ssu18_edaphic_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")edaphic_sig_diff<-base::union(edaphic_md_signif$parameters, row.names(summary(ssu18_edaphic_bioenv_ind_mantel)))new_edaphic_md_signif_hits<-edaphic_sig_diffedaphic_md_signif_all<-edaphic_md_scores[edaphic_md_scores$parameters%in%new_edaphic_md_signif_hits,]## Check. Next, we run `envfit` for the ASVs.envfit_edaphic_asv<-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10], perm =1000, choices =c(1, 2))edaphic_asv_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="species")edaphic_asv_scores<-edaphic_asv_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")edaphic_asv_scores[,1]<-NULLedaphic_asv_signif_hits<-base::subset(envfit_edaphic_asv$vectors$pvals, c(envfit_edaphic_asv$vectors$pvals<0.05&envfit_edaphic_asv$vectors$r>0.5))edaphic_asv_signif_hits<-data.frame(edaphic_asv_signif_hits)edaphic_asv_signif_hits<-rownames(edaphic_asv_signif_hits)edaphic_asv_signif<-edaphic_asv_scores[edaphic_asv_scores$parameters%in%edaphic_asv_signif_hits,]edaphic_md_signif_all$variable_type<-"metadata"edaphic_asv_signif$variable_type<-"ASV"edaphic_bioplot_data<-rbind(edaphic_md_signif_all, edaphic_asv_signif)## The last thing to do is categorize parameters scores and ASV ## scores into different variable types for plotting.edaphic_bioplot_data_md<-subset(edaphic_bioplot_data, edaphic_bioplot_data$variable_type=="metadata")edaphic_bioplot_data_asv<-subset(edaphic_bioplot_data, edaphic_bioplot_data$variable_type=="ASV")## code for the plotedaphic_cap_vals<-data.frame(edaphic_cap$CCA$eig[1:2])edaphic_cap1<-signif((edaphic_cap_vals[1,]*100), digits =3)edaphic_cap2<-signif((edaphic_cap_vals[2,]*100), digits =3)cpa1_lab<-paste("CAP1", " (", edaphic_cap1, "%)", sep ="")cpa2_lab<-paste("CAP2", " (", edaphic_cap2, "%)", sep ="")swel_col<-c("#2271B2", "#71B222", "#B22271")edaphic_plot<-ggplot(edaphic_plot_data)+geom_point(mapping =aes(x =CAP1, y =CAP2, colour =TREAT_T), size =8)+scale_colour_manual(values =swel_col)+geom_segment(aes(x =0, y =0, xend =CAP1, yend =CAP2), data =edaphic_bioplot_data_md, linetype ="solid", arrow =arrow(length =unit(0.3, "cm")), size =0.9, color ="#191919", inherit.aes =FALSE)+geom_text(data =edaphic_bioplot_data_md, aes(x =CAP1, y =CAP2, label =parameters), size =7, nudge_x =0.1, nudge_y =0.05)+theme_classic(base_size =12)+labs(subtitle ="Edaphic properties", x =cpa1_lab, y =cpa2_lab)edaphic_plot<-edaphic_plot+coord_fixed()+theme(aspect.ratio =1)rm(list =ls(pattern ="tmp_"))########################################################################### 16S rRNA Soil Functional Response ###########################################################################tmp_md<-ssu18_select_mc_norm_split_no_ac$soil_functtmp_md$TREAT_T<-as.character(tmp_md$TREAT_T)tmp_comm<-data.frame(t(ssu18_select_mc_norm_split_no_ac$data_loaded))soil_funct_rank<-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow","bra", "kul"), stepacross =FALSE, method ="spearman")## Let's run `capscale` using Bray-Curtis. ## * Starting properties: micC, micN, micP, micCN, micCP, micNP, AG_ase, BG_ase, BP_ase, CE_ase, P_ase, N_ase, S_ase, XY_ase, LP_ase, PX_ase, CO2, enzCN, enzCP, enzNP## * Autocorrelated removed: micN, micNP, enzCN, enzCP, BP_ase, CE_ase, LP_ase, N_ase, P_ase## * Remove for capscale: NONEsoil_funct_cap<-capscale(tmp_comm~micC+micP+micCN+micCP+AG_ase+BG_ase+S_ase+XY_ase+PX_ase+CO2+enzNP, tmp_md, dist ="bray")tmp_auto_plt<-autoplot(soil_funct_cap, arrows =TRUE)anova(soil_funct_cap)# overall test of the significant of the analysisanova(soil_funct_cap, by ="axis", perm.max =500)# test axes for significanceanova(soil_funct_cap, by ="terms", permu =500)# test for sign. environ. variablestmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")soil_funct_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")soil_funct_md_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="biplot")soil_funct_md_scores[,1]<-NULLsoil_funct_md_scores<-soil_funct_md_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")tmp_samp_scores_sub<-soil_funct_plot_data[, 6:7]tmp_samp_scores_sub<-as.matrix(tmp_samp_scores_sub)tmp_param_list<-soil_funct_md_scores$parameterstmp_md_sub<-subset(tmp_md, select =tmp_param_list)envfit_soil_funct_md<-envfit(tmp_samp_scores_sub, tmp_md_sub, perm =1000, choices =c(1, 2))soil_funct_md_signif_hits<-base::subset(envfit_soil_funct_md$vectors$pvals, c(envfit_soil_funct_md$vectors$pvals<0.05&envfit_soil_funct_md$vectors$r>0.4))soil_funct_md_signif_hits<-data.frame(soil_funct_md_signif_hits)soil_funct_md_signif_hits<-rownames(soil_funct_md_signif_hits)soil_funct_md_signif<-soil_funct_md_scores[soil_funct_md_scores$parameters%in%soil_funct_md_signif_hits,]soil_funct_md_signif$parametersprint("Significant parameters from bioenv analysis.")row.names(summary(ssu18_soil_funct_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")soil_funct_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(ssu18_soil_funct_bioenv_ind_mantel)), soil_funct_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(soil_funct_md_signif$parameters, row.names(summary(ssu18_soil_funct_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")soil_funct_sig_diff<-base::union(soil_funct_md_signif$parameters, row.names(summary(ssu18_soil_funct_bioenv_ind_mantel)))soil_funct_sig_diffnew_soil_funct_md_signif_hits<-soil_funct_sig_diffsoil_funct_md_signif_all<-soil_funct_md_scores[soil_funct_md_scores$parameters%in%new_soil_funct_md_signif_hits,]envfit_soil_funct_asv<-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10], perm =1000, choices =c(1, 2))soil_funct_asv_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="species")soil_funct_asv_scores<-soil_funct_asv_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")soil_funct_asv_scores[,1]<-NULLsoil_funct_asv_signif_hits<-base::subset(envfit_soil_funct_asv$vectors$pvals, c(envfit_soil_funct_asv$vectors$pvals<0.05&envfit_soil_funct_asv$vectors$r>0.5))soil_funct_asv_signif_hits<-data.frame(soil_funct_asv_signif_hits)soil_funct_asv_signif_hits<-rownames(soil_funct_asv_signif_hits)soil_funct_asv_signif<-soil_funct_asv_scores[soil_funct_asv_scores$parameters%in%soil_funct_asv_signif_hits,]soil_funct_md_signif_all$variable_type<-"metadata"soil_funct_asv_signif$variable_type<-"ASV"soil_funct_bioplot_data<-rbind(soil_funct_md_signif_all, soil_funct_asv_signif)soil_funct_bioplot_data_md<-subset(soil_funct_bioplot_data, soil_funct_bioplot_data$variable_type=="metadata")soil_funct_bioplot_data_asv<-subset(soil_funct_bioplot_data, soil_funct_bioplot_data$variable_type=="ASV")## PLOT Codesoil_funct_cap_vals<-data.frame(soil_funct_cap$CCA$eig[1:2])soil_funct_cap1<-signif((soil_funct_cap_vals[1,]*100), digits =3)soil_funct_cap2<-signif((soil_funct_cap_vals[2,]*100), digits =3)cpa1_lab<-paste("CAP1", " (", soil_funct_cap1, "%)", sep ="")cpa2_lab<-paste("CAP2", " (", soil_funct_cap2, "%)", sep ="")swel_col<-c("#2271B2", "#71B222", "#B22271")soil_funct_plot<-ggplot(soil_funct_plot_data)+geom_point(mapping =aes(x =CAP1, y =CAP2, colour =TREAT_T), size =8)+scale_colour_manual(values =swel_col)+geom_segment(aes(x =0, y =0, xend =CAP1, yend =CAP2), data =soil_funct_bioplot_data_md, linetype ="solid", arrow =arrow(length =unit(0.3, "cm")), size =0.9, color ="#191919")+geom_text(data =soil_funct_bioplot_data_md, aes(x =CAP1, y =CAP2, label =parameters), size =7, nudge_x =0.1, nudge_y =0.05)+theme_classic(base_size =12)+labs(subtitle ="Functional Response", x =cpa1_lab, y =cpa2_lab)soil_funct_plot<-soil_funct_plot+coord_fixed()+theme(aspect.ratio =1)rm(list =ls(pattern ="tmp_"))############################################################################# 16S rRNA Temperature Adaptation ############################################################################## tmp_md<-ssu18_select_mc_norm_split_no_ac$temp_adapttmp_md$TREAT_T<-as.character(tmp_md$TREAT_T)tmp_comm<-data.frame(t(ssu18_select_mc_norm_split_no_ac$data_loaded))temp_adapt_rank<-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow","bra", "kul"), stepacross =FALSE, method ="spearman")## Let's run `capscale` using Bray-Curtis. ## * Starting properties: AG_Q10, BG_Q10, BP_Q10, CE_Q10, P_Q10, N_Q10, S_Q10, XY_Q10, LP_Q10, PX_Q10, CUEcn, CUEcp, NUE, PUE, Tmin, SI## * Autocorrelated removed: NUE, PUE, SI## * Remove for capscale: NONEtemp_adapt_cap<-capscale(tmp_comm~AG_Q10+BG_Q10+BP_Q10+CE_Q10+P_Q10+N_Q10+S_Q10+XY_Q10+LP_Q10+PX_Q10+CUEcn+CUEcp+Tmin, tmp_md, dist ="bray")tmp_auto_plt<-autoplot(temp_adapt_cap, arrows =TRUE)anova(temp_adapt_cap)# overall test of the significant of the analysisanova(temp_adapt_cap, by ="axis", perm.max =500)# test axes for significanceanova(temp_adapt_cap, by ="terms", permu =500)# test for sign. environ. variablestmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")temp_adapt_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")temp_adapt_md_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="biplot")temp_adapt_md_scores[,1]<-NULLtemp_adapt_md_scores<-temp_adapt_md_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")tmp_samp_scores_sub<-temp_adapt_plot_data[, 6:7]tmp_samp_scores_sub<-as.matrix(tmp_samp_scores_sub)tmp_param_list<-temp_adapt_md_scores$parameterstmp_md_sub<-subset(tmp_md, select =tmp_param_list)envfit_temp_adapt_md<-envfit(tmp_samp_scores_sub, tmp_md_sub, perm =1000, choices =c(1, 2))temp_adapt_md_signif_hits<-base::subset(envfit_temp_adapt_md$vectors$pvals, c(envfit_temp_adapt_md$vectors$pvals<0.05&envfit_temp_adapt_md$vectors$r>0.4))temp_adapt_md_signif_hits<-data.frame(temp_adapt_md_signif_hits)temp_adapt_md_signif_hits<-rownames(temp_adapt_md_signif_hits)temp_adapt_md_signif<-temp_adapt_md_scores[temp_adapt_md_scores$parameters%in%temp_adapt_md_signif_hits,]print("Significant parameters from bioenv analysis.")row.names(summary(ssu18_temp_adapt_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")temp_adapt_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(ssu18_temp_adapt_bioenv_ind_mantel)), temp_adapt_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(temp_adapt_md_signif$parameters, row.names(summary(ssu18_temp_adapt_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")temp_adapt_sig_diff<-base::union(temp_adapt_md_signif$parameters, row.names(summary(ssu18_temp_adapt_bioenv_ind_mantel)))temp_adapt_sig_diffnew_temp_adapt_md_signif_hits<-temp_adapt_sig_difftemp_adapt_md_signif_all<-temp_adapt_md_scores[temp_adapt_md_scores$parameters%in%new_temp_adapt_md_signif_hits,]envfit_temp_adapt_asv<-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10], perm =1000, choices =c(1, 2))temp_adapt_asv_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="species")temp_adapt_asv_scores<-temp_adapt_asv_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")temp_adapt_asv_scores[,1]<-NULLtemp_adapt_asv_signif_hits<-base::subset(envfit_temp_adapt_asv$vectors$pvals, c(envfit_temp_adapt_asv$vectors$pvals<0.05&envfit_temp_adapt_asv$vectors$r>0.5))temp_adapt_asv_signif_hits<-data.frame(temp_adapt_asv_signif_hits)temp_adapt_asv_signif_hits<-rownames(temp_adapt_asv_signif_hits)temp_adapt_asv_signif<-temp_adapt_asv_scores[temp_adapt_asv_scores$parameters%in%temp_adapt_asv_signif_hits,]temp_adapt_md_signif_all$variable_type<-"metadata"temp_adapt_asv_signif$variable_type<-"ASV"temp_adapt_bioplot_data<-rbind(temp_adapt_md_signif_all, temp_adapt_asv_signif)temp_adapt_bioplot_data_md<-subset(temp_adapt_bioplot_data, temp_adapt_bioplot_data$variable_type=="metadata")temp_adapt_bioplot_data_asv<-subset(temp_adapt_bioplot_data, temp_adapt_bioplot_data$variable_type=="ASV")temp_adapt_cap_vals<-data.frame(temp_adapt_cap$CCA$eig[1:2])temp_adapt_cap1<-signif((temp_adapt_cap_vals[1,]*100), digits =3)temp_adapt_cap2<-signif((temp_adapt_cap_vals[2,]*100), digits =3)cpa1_lab<-paste("CAP1", " (", temp_adapt_cap1, "%)", sep ="")cpa2_lab<-paste("CAP2", " (", temp_adapt_cap2, "%)", sep ="")swel_col<-c("#2271B2", "#71B222", "#B22271")temp_adapt_plot<-ggplot(temp_adapt_plot_data)+geom_point(mapping =aes(x =CAP1, y =CAP2, colour =TREAT_T), size =8)+scale_colour_manual(values =swel_col)+geom_segment(aes(x =0, y =0, xend =CAP1, yend =CAP2), data =temp_adapt_bioplot_data_md, linetype ="solid", arrow =arrow(length =unit(0.3, "cm")), size =0.9, color ="#191919", inherit.aes =FALSE)+geom_text(data =temp_adapt_bioplot_data_md, aes(x =CAP1, y =CAP2, label =parameters), size =7, nudge_x =0.1, nudge_y =0.05)+theme_classic(base_size =12)+labs(subtitle ="Temperature Adaptation", x =cpa1_lab, y =cpa2_lab)temp_adapt_plot<-temp_adapt_plot+coord_fixed()+theme(aspect.ratio=1)objects(pattern ="_plot")ssu18_edaphic_plot<-edaphic_plotssu18_soil_funct_plot<-soil_funct_plotssu18_temp_adapt_plot<-temp_adapt_plotgdata::keep(ssu18_edaphic_plot, ssu18_soil_funct_plot, ssu18_temp_adapt_plot, its18_edaphic_bioenv_ind_mantel, its18_select_mc_norm_split_no_ac, its18_soil_funct_bioenv_ind_mantel, its18_temp_adapt_bioenv_ind_mantel, ssu18_edaphic_bioenv_ind_mantel, ssu18_select_mc_norm_split_no_ac, ssu18_soil_funct_bioenv_ind_mantel, ssu18_temp_adapt_bioenv_ind_mantel, sure =TRUE)################################################################################ ITS EDAPHIC PROPERTIES #################################################################################tmp_md<-its18_select_mc_norm_split_no_ac$edaphictmp_md$TREAT_T<-as.character(tmp_md$TREAT_T)tmp_comm<-data.frame(t(its18_select_mc_norm_split_no_ac$data_loaded))edaphic_rank<-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow","bra", "kul"), stepacross =FALSE, method ="spearman")## Run `capscale` using Bray-Curtis. ## * Starting properties: AST, H2O, N, P, Al, Ca, Fe, K, Mg, Mn, Na, TEB, ECEC, pH, NH4, NO3, PO4, DOC, DON, DOCN.## * Autocorrelated removed: TEB, DON, Na, Al, Ca.## * Remove for capscale: Mg, Mn, Na, Al, Fe, Kedaphic_cap<-capscale(tmp_comm~AST+H2O+N+P+ECEC+pH+NH4+NO3+PO4+DOC+DOCN, tmp_md, dist ="bray")anova(edaphic_cap)# overall test of the significant of the analysisanova(edaphic_cap, by ="axis", perm.max =500)# test axes for significanceanova(edaphic_cap, by ="terms", permu =500)# test for sign. environ. variables## Next, we need to grab capscale scores for the samples and create a ## data frame of the first two dimensions. We will also need to add some ## of the sample details to the data frame. For this we use the vegan ## function `scores` which gets species or site scores from the ordination.library(ggvegan)tmp_auto_plt<-ggplot2::autoplot(edaphic_cap, arrows =TRUE)tmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")edaphic_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")## Now we have a new data frame that contains sample details and capscale values. ## We can then do the same with the metadata vectors. ## Here though we only need the scores and parameter name. edaphic_md_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="biplot")edaphic_md_scores[,1]<-NULLedaphic_md_scores<-edaphic_md_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")## Let's run some quick correlations of metadata with ordination axes to ## see which parameters are significant. For this we use the vegan function `envfit`.tmp_samp_scores_sub<-edaphic_plot_data[, 6:7]tmp_samp_scores_sub<-as.matrix(tmp_samp_scores_sub)tmp_param_list<-edaphic_md_scores$parameterstmp_md_sub<-subset(tmp_md, select =tmp_param_list)envfit_edaphic_md<-envfit(tmp_samp_scores_sub, tmp_md_sub, perm =1000, choices =c(1, 2))edaphic_md_signif_hits<-base::subset(envfit_edaphic_md$vectors$pvals, c(envfit_edaphic_md$vectors$pvals<0.05&envfit_edaphic_md$vectors$r>0.4))edaphic_md_signif_hits<-data.frame(edaphic_md_signif_hits)edaphic_md_signif_hits<-rownames(edaphic_md_signif_hits)edaphic_md_signif<-edaphic_md_scores[edaphic_md_scores$parameters%in%edaphic_md_signif_hits,]edaphic_md_signif$parameters## Now let's see if the same parameters are significant for the `envfit` and `bioenv` analyses.print("Significant parameters from bioenv analysis.")row.names(summary(its18_edaphic_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")edaphic_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(its18_edaphic_bioenv_ind_mantel)), edaphic_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(edaphic_md_signif$parameters, row.names(summary(its18_edaphic_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")edaphic_sig_diff<-base::union(edaphic_md_signif$parameters, row.names(summary(its18_edaphic_bioenv_ind_mantel)))edaphic_sig_diffnew_edaphic_md_signif_hits<-edaphic_sig_diffedaphic_md_signif_all<-edaphic_md_scores[edaphic_md_scores$parameters%in%new_edaphic_md_signif_hits,]## Check. Next, we run `envfit` for the ASVs.envfit_edaphic_asv<-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10], perm =1000, choices =c(1, 2))edaphic_asv_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="species")edaphic_asv_scores<-edaphic_asv_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")edaphic_asv_scores[,1]<-NULLedaphic_asv_signif_hits<-base::subset(envfit_edaphic_asv$vectors$pvals, c(envfit_edaphic_asv$vectors$pvals<0.05&envfit_edaphic_asv$vectors$r>0.5))edaphic_asv_signif_hits<-data.frame(edaphic_asv_signif_hits)edaphic_asv_signif_hits<-rownames(edaphic_asv_signif_hits)edaphic_asv_signif<-edaphic_asv_scores[edaphic_asv_scores$parameters%in%edaphic_asv_signif_hits,]edaphic_md_signif_all$variable_type<-"metadata"edaphic_asv_signif$variable_type<-"ASV"edaphic_bioplot_data<-rbind(edaphic_md_signif_all, edaphic_asv_signif)## The last thing to do is categorize parameters scores and ASV ## scores into different variable types for plotting.edaphic_bioplot_data_md<-subset(edaphic_bioplot_data, edaphic_bioplot_data$variable_type=="metadata")edaphic_bioplot_data_asv<-subset(edaphic_bioplot_data, edaphic_bioplot_data$variable_type=="ASV")## code for the plotedaphic_cap_vals<-data.frame(edaphic_cap$CCA$eig[1:2])edaphic_cap1<-signif((edaphic_cap_vals[1,]*100), digits =3)edaphic_cap2<-signif((edaphic_cap_vals[2,]*100), digits =3)cpa1_lab<-paste("CAP1", " (", edaphic_cap1, "%)", sep ="")cpa2_lab<-paste("CAP2", " (", edaphic_cap2, "%)", sep ="")swel_col<-c("#2271B2", "#71B222", "#B22271")edaphic_plot<-ggplot(edaphic_plot_data)+geom_point(mapping =aes(x =CAP1, y =CAP2, colour =TREAT_T), size =8)+scale_colour_manual(values =swel_col)+geom_segment(aes(x =0, y =0, xend =CAP1, yend =CAP2), data =edaphic_bioplot_data_md, linetype ="solid", arrow =arrow(length =unit(0.3, "cm")), size =0.9, color ="#191919", inherit.aes =FALSE)+geom_text(data =edaphic_bioplot_data_md, aes(x =CAP1, y =CAP2, label =parameters), size =7, nudge_x =0.1, nudge_y =0.05)+theme_classic(base_size =12)+labs(x =cpa1_lab, y =cpa2_lab)edaphic_plot<-edaphic_plot+coord_fixed()+theme(aspect.ratio =1)edaphic_plotrm(list =ls(pattern ="tmp_"))################################################################################ ITS Soil Functional Response ###########################################################################tmp_md<-its18_select_mc_norm_split_no_ac$soil_functtmp_md$TREAT_T<-as.character(tmp_md$TREAT_T)tmp_comm<-data.frame(t(its18_select_mc_norm_split_no_ac$data_loaded))soil_funct_rank<-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow", "bra", "kul"), stepacross =FALSE, method ="spearman")## Let's run `capscale` using Bray-Curtis ## * Starting properties: micC, micN, micP, micCN, micCP, micNP, AG_ase, BG_ase, BP_ase, CE_ase, P_ase, N_ase, S_ase, XY_ase, LP_ase, PX_ase, CO2, enzCN, enzCP, enzNP## * Autocorrelated removed: micN, micNP, enzCN, enzCP, BP_ase, CE_ase, LP_ase, N_ase, P_ase## * Remove for capscale: NONEsoil_funct_cap<-capscale(tmp_comm~micC+micP+micCN+micCP+AG_ase+BG_ase+S_ase+XY_ase+PX_ase+CO2+enzNP, tmp_md, dist ="bray")tmp_auto_plt<-autoplot(soil_funct_cap, arrows =TRUE)anova(soil_funct_cap)# overall test of the significant of the analysisanova(soil_funct_cap, by ="axis", perm.max =500)# test axes for significanceanova(soil_funct_cap, by ="terms", permu =500)# test for sign. environ. variablestmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")soil_funct_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")soil_funct_md_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="biplot")soil_funct_md_scores[,1]<-NULLsoil_funct_md_scores<-soil_funct_md_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")tmp_samp_scores_sub<-soil_funct_plot_data[, 6:7]tmp_samp_scores_sub<-as.matrix(tmp_samp_scores_sub)tmp_param_list<-soil_funct_md_scores$parameterstmp_md_sub<-subset(tmp_md, select =tmp_param_list)envfit_soil_funct_md<-envfit(tmp_samp_scores_sub, tmp_md_sub, perm =1000, choices =c(1, 2))soil_funct_md_signif_hits<-base::subset(envfit_soil_funct_md$vectors$pvals, c(envfit_soil_funct_md$vectors$pvals<0.05&envfit_soil_funct_md$vectors$r>0.4))soil_funct_md_signif_hits<-data.frame(soil_funct_md_signif_hits)soil_funct_md_signif_hits<-rownames(soil_funct_md_signif_hits)soil_funct_md_signif<-soil_funct_md_scores[soil_funct_md_scores$parameters%in%soil_funct_md_signif_hits,]soil_funct_md_signif$parametersprint("Significant parameters from bioenv analysis.")row.names(summary(its18_soil_funct_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")soil_funct_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(its18_soil_funct_bioenv_ind_mantel)), soil_funct_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(soil_funct_md_signif$parameters, row.names(summary(its18_soil_funct_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")soil_funct_sig_diff<-base::union(soil_funct_md_signif$parameters, row.names(summary(its18_soil_funct_bioenv_ind_mantel)))soil_funct_sig_diffnew_soil_funct_md_signif_hits<-soil_funct_sig_diffsoil_funct_md_signif_all<-soil_funct_md_scores[soil_funct_md_scores$parameters%in%new_soil_funct_md_signif_hits,]envfit_soil_funct_asv<-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10], perm =1000, choices =c(1, 2))soil_funct_asv_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="species")soil_funct_asv_scores<-soil_funct_asv_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")soil_funct_asv_scores[,1]<-NULLsoil_funct_asv_signif_hits<-base::subset(envfit_soil_funct_asv$vectors$pvals, c(envfit_soil_funct_asv$vectors$pvals<0.05&envfit_soil_funct_asv$vectors$r>0.5))soil_funct_asv_signif_hits<-data.frame(soil_funct_asv_signif_hits)soil_funct_asv_signif_hits<-rownames(soil_funct_asv_signif_hits)soil_funct_asv_signif<-soil_funct_asv_scores[soil_funct_asv_scores$parameters%in%soil_funct_asv_signif_hits,]soil_funct_md_signif_all$variable_type<-"metadata"soil_funct_asv_signif$variable_type<-"ASV"soil_funct_bioplot_data<-rbind(soil_funct_md_signif_all, soil_funct_asv_signif)soil_funct_bioplot_data_md<-subset(soil_funct_bioplot_data, soil_funct_bioplot_data$variable_type=="metadata")soil_funct_bioplot_data_asv<-subset(soil_funct_bioplot_data, soil_funct_bioplot_data$variable_type=="ASV")## PLOT Codesoil_funct_cap_vals<-data.frame(soil_funct_cap$CCA$eig[1:2])soil_funct_cap1<-signif((soil_funct_cap_vals[1,]*100), digits =3)soil_funct_cap2<-signif((soil_funct_cap_vals[2,]*100), digits =3)cpa1_lab<-paste("CAP1", " (", soil_funct_cap1, "%)", sep ="")cpa2_lab<-paste("CAP2", " (", soil_funct_cap2, "%)", sep ="")swel_col<-c("#2271B2", "#71B222", "#B22271")soil_funct_plot<-ggplot(soil_funct_plot_data)+geom_point(mapping =aes(x =CAP1, y =CAP2, colour =TREAT_T), size =8)+scale_colour_manual(values =swel_col)+geom_segment(aes(x =0, y =0, xend =CAP1, yend =CAP2), data =soil_funct_bioplot_data_md, linetype ="solid", arrow =arrow(length =unit(0.3, "cm")), size =0.9, color ="#191919")+geom_text(data =soil_funct_bioplot_data_md, aes(x =CAP1, y =CAP2, label =parameters), size =7, nudge_x =0.1, nudge_y =0.05)+theme_classic(base_size =12)+labs(x =cpa1_lab, y =cpa2_lab)soil_funct_plot<-soil_funct_plot+coord_fixed()+theme(aspect.ratio =1)rm(list =ls(pattern ="tmp_"))################################################################################ ITS Temperature Adaptation #############################################################################tmp_md<-its18_select_mc_norm_split_no_ac$temp_adapttmp_md$TREAT_T<-as.character(tmp_md$TREAT_T)tmp_comm<-data.frame(t(its18_select_mc_norm_split_no_ac$data_loaded))temp_adapt_rank<-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow", "bra", "kul"), stepacross =FALSE, method ="spearman")## Let's run `capscale` using Bray-Curtis. ## * Starting properties: AG_Q10, BG_Q10, BP_Q10, CE_Q10, P_Q10, N_Q10, S_Q10, XY_Q10, LP_Q10, PX_Q10, CUEcn, CUEcp, NUE, PUE, Tmin, SI## * Autocorrelated removed: NUE, PUE, P_Q10, SI ## * Remove for capscale: S_Q10temp_adapt_cap<-capscale(tmp_comm~AG_Q10+BG_Q10+BP_Q10+CE_Q10+N_Q10+XY_Q10+LP_Q10+PX_Q10+CUEcn+CUEcp+Tmin, tmp_md, dist ="bray")tmp_auto_plt<-autoplot(temp_adapt_cap, arrows =TRUE)anova(temp_adapt_cap)# overall test of the significant of the analysisanova(temp_adapt_cap, by ="axis", perm.max =500)# test axes for significanceanova(temp_adapt_cap, by ="terms", permu =500)# test for sign. environ. variablestmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")temp_adapt_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")temp_adapt_md_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="biplot")temp_adapt_md_scores[,1]<-NULLtemp_adapt_md_scores<-temp_adapt_md_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")tmp_samp_scores_sub<-temp_adapt_plot_data[, 6:7]tmp_samp_scores_sub<-as.matrix(tmp_samp_scores_sub)tmp_param_list<-temp_adapt_md_scores$parameterstmp_md_sub<-subset(tmp_md, select =tmp_param_list)envfit_temp_adapt_md<-envfit(tmp_samp_scores_sub, tmp_md_sub, perm =1000, choices =c(1, 2))temp_adapt_md_signif_hits<-base::subset(envfit_temp_adapt_md$vectors$pvals, c(envfit_temp_adapt_md$vectors$pvals<0.05&envfit_temp_adapt_md$vectors$r>0.4))temp_adapt_md_signif_hits<-data.frame(temp_adapt_md_signif_hits)temp_adapt_md_signif_hits<-rownames(temp_adapt_md_signif_hits)temp_adapt_md_signif<-temp_adapt_md_scores[temp_adapt_md_scores$parameters%in%temp_adapt_md_signif_hits,]print("Significant parameters from bioenv analysis.")row.names(summary(its18_temp_adapt_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")temp_adapt_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(its18_temp_adapt_bioenv_ind_mantel)), temp_adapt_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(temp_adapt_md_signif$parameters, row.names(summary(its18_temp_adapt_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")temp_adapt_sig_diff<-base::union(temp_adapt_md_signif$parameters, row.names(summary(its18_temp_adapt_bioenv_ind_mantel)))temp_adapt_sig_diffnew_temp_adapt_md_signif_hits<-temp_adapt_sig_diff[1:4]temp_adapt_md_signif_all<-temp_adapt_md_scores[temp_adapt_md_scores$parameters%in%new_temp_adapt_md_signif_hits,]envfit_temp_adapt_asv<-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10], perm =1000, choices =c(1, 2))temp_adapt_asv_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="species")temp_adapt_asv_scores<-temp_adapt_asv_scores%>%dplyr::mutate(parameters =Label, .before =CAP1)%>%tibble::column_to_rownames("Label")temp_adapt_asv_scores[,1]<-NULLtemp_adapt_asv_signif_hits<-base::subset(envfit_temp_adapt_asv$vectors$pvals, c(envfit_temp_adapt_asv$vectors$pvals<0.05&envfit_temp_adapt_asv$vectors$r>0.5))temp_adapt_asv_signif_hits<-data.frame(temp_adapt_asv_signif_hits)temp_adapt_asv_signif_hits<-rownames(temp_adapt_asv_signif_hits)temp_adapt_asv_signif<-temp_adapt_asv_scores[temp_adapt_asv_scores$parameters%in%temp_adapt_asv_signif_hits,]temp_adapt_md_signif_all$variable_type<-"metadata"temp_adapt_asv_signif$variable_type<-"ASV"temp_adapt_bioplot_data<-rbind(temp_adapt_md_signif_all, temp_adapt_asv_signif)temp_adapt_bioplot_data_md<-subset(temp_adapt_bioplot_data, temp_adapt_bioplot_data$variable_type=="metadata")temp_adapt_bioplot_data_asv<-subset(temp_adapt_bioplot_data, temp_adapt_bioplot_data$variable_type=="ASV")temp_adapt_cap_vals<-data.frame(temp_adapt_cap$CCA$eig[1:2])temp_adapt_cap1<-signif((temp_adapt_cap_vals[1,]*100), digits =3)temp_adapt_cap2<-signif((temp_adapt_cap_vals[2,]*100), digits =3)cpa1_lab<-paste("CAP1", " (", temp_adapt_cap1, "%)", sep ="")cpa2_lab<-paste("CAP2", " (", temp_adapt_cap2, "%)", sep ="")swel_col<-c("#2271B2", "#71B222", "#B22271")temp_adapt_plot<-ggplot(temp_adapt_plot_data)+geom_point(mapping =aes(x =CAP1, y =CAP2, colour =TREAT_T), size =8)+scale_colour_manual(values =swel_col)+geom_segment(aes(x =0, y =0, xend =CAP1, yend =CAP2), data =temp_adapt_bioplot_data_md, linetype ="solid", arrow =arrow(length =unit(0.3, "cm")), size =0.9, color ="#191919", inherit.aes =FALSE)+geom_text(data =temp_adapt_bioplot_data_md, aes(x =CAP1, y =CAP2, label =parameters), size =7, nudge_x =0.1, nudge_y =0.05)+theme_classic(base_size =12)+labs(x =cpa1_lab, y =cpa2_lab)temp_adapt_plot<-temp_adapt_plot+coord_fixed()+theme(aspect.ratio =1)its18_edaphic_plot<-edaphic_plotits18_soil_funct_plot<-soil_funct_plotits18_temp_adapt_plot<-temp_adapt_plot### BUILD the final Plottmp_plot_final<-(ssu18_edaphic_plot|ssu18_soil_funct_plot)/(its18_edaphic_plot|its18_soil_funct_plot)+patchwork::plot_annotation(tag_levels ="a", title =NULL, subtitle =NULL, caption =NULL)tmp_plot_final<-tmp_plot_final+patchwork::plot_layout(guides ="collect")&theme(legend.position ="bottom", plot.title =element_text(size =24), plot.tag =element_text(size =31), axis.title =element_text(size =18), axis.text =element_text(size =16))ggplot2::ggsave(tmp_plot_final, path ="include/pub/EXD/EXD_Figure_5/", filename ="Extended_Data_Figure_5.png", height =8398, width =7485, units ='px', dpi =600, bg ="white")ggplot2::ggsave(tmp_plot_final, path ="include/pub/EXD/EXD_Figure_5/", filename ="Extended_Data_Figure_5.pdf", height =8398, width =7485, units ='px', dpi =600, bg ="white")
## Load dataremove(list =ls())set.seed(119)ssu18_ps_work<-readRDS("include/pub/SOM/Supplementary_Figure_1.rds")## 1) Get all Class-level Proteobacteria namesssu18_data_sets<-c("ssu18_ps_work")for(iinssu18_data_sets){tmp_name<-purrr::map_chr(i, ~paste0(., "_proteo"))tmp_get<-get(i)tmp_df<-subset_taxa(tmp_get, Phylum=="Proteobacteria")assign(tmp_name, tmp_df)print(tmp_name)tmp_get_taxa<-get_taxa_unique(tmp_df, taxonomic.rank =rank_names(tmp_df)[3], errorIfNULL =TRUE)print(tmp_get_taxa)rm(list =ls(pattern ="tmp_"))rm(list =ls(pattern ="_proteo"))}## 2) Replace Phylum Proteobacteria with the Class name.for(jinssu18_data_sets){tmp_name<-purrr::map_chr(j, ~paste0(., "_proteo_clean"))tmp_get<-get(j)tmp_clean<-data.frame(tax_table(tmp_get))for(iin1:nrow(tmp_clean)){if(tmp_clean[i, 2]=="Proteobacteria"&tmp_clean[i, 3]=="Alphaproteobacteria"){phylum<-base::paste("Alphaproteobacteria")tmp_clean[i, 2]<-phylum}elseif(tmp_clean[i, 2]=="Proteobacteria"&tmp_clean[i, 3]=="Gammaproteobacteria"){phylum<-base::paste("Gammaproteobacteria")tmp_clean[i, 2]<-phylum}elseif(tmp_clean[i, 2]=="Proteobacteria"&tmp_clean[i, 3]=="Zetaproteobacteria"){phylum<-base::paste("Zetaproteobacteria")tmp_clean[i, 2]<-phylum}elseif(tmp_clean[i, 2]=="Proteobacteria"&tmp_clean[i, 3]=="p_Proteobacteria"){phylum<-base::paste("p_Proteobacteria")tmp_clean[i, 2]<-phylum}}tax_table(tmp_get)<-as.matrix(tmp_clean)rank_names(tmp_get)assign(tmp_name, tmp_get)print(c(tmp_name, tmp_get))print(length(get_taxa_unique(tmp_get, taxonomic.rank =rank_names(tmp_get)[2], errorIfNULL =TRUE)))tmp_path<-file.path("include/pub/SOM/")rm(list =ls(pattern ="tmp_"))}rm(class, order, phylum)## 3) In order to use `microeco`, we need to add the rank designation as a prefix to each taxa. ## For example, `Actinobacteriota` is changed to `p__Actinobacteriota`. for(iinssu18_data_sets){tmp_get<-get(purrr::map_chr(i, ~paste0(., "_proteo_clean")))tmp_sam_data<-sample_data(tmp_get)tmp_tax_data<-data.frame(tax_table(tmp_get))tmp_tax_data$Phylum<-gsub("p_Proteobacteria", "Proteobacteria", tmp_tax_data$Phylum)tmp_tax_data$ASV_ID<-NULL# Some have, some do nottmp_tax_data$ASV_SEQ<-NULLtmp_tax_data[]<-data.frame(lapply(tmp_tax_data,gsub, pattern ="^[k | p | c | o | f]_.*", replacement ="", fixed =FALSE))tmp_tax_data$Kingdom<-paste("k__", tmp_tax_data$Kingdom, sep ="")tmp_tax_data$Phylum<-paste("p__", tmp_tax_data$Phylum, sep ="")tmp_tax_data$Class<-paste("c__", tmp_tax_data$Class, sep ="")tmp_tax_data$Order<-paste("o__", tmp_tax_data$Order, sep ="")tmp_tax_data$Family<-paste("f__", tmp_tax_data$Family, sep ="")tmp_tax_data$Genus<-paste("g__", tmp_tax_data$Genus, sep ="")tmp_tax_data<-as.matrix(tmp_tax_data)tmp_ps<-phyloseq(otu_table(tmp_get),phy_tree(tmp_get),tax_table(tmp_tax_data),tmp_sam_data)assign(i, tmp_ps)rm(list =ls(pattern ="tmp_"))}rm(list =ls(pattern ="_proteo_clean"))## 4) Next, we need to covert each `phyloseq object` to a `microtable class`. ## The microtable class is the basic data structure for the `microeco` package ## and designed to store basic information from all the downstream analyses ## (e.g, alpha diversity, beta diversity, etc.). ## We use the [file2meco](https://github.com/ChiLiubio/file2meco) to read the phyloseq ## object and convert into a microtable object. We can add `_me` as a suffix ## to each object to distiguish it from its' phyloseq counterpart. for(iinssu18_data_sets){tmp_get<-get(i)tmp_otu_table<-data.frame(t(otu_table(tmp_get)))tmp_sample_info<-data.frame(sample_data(tmp_get))tmp_taxonomy_table<-data.frame(tax_table(tmp_get))tmp_phylo_tree<-phy_tree(tmp_get)tmp_taxonomy_table%<>%tidy_taxonomytmp_dataset<-microtable$new( sample_table =tmp_sample_info, otu_table =tmp_otu_table, tax_table =tmp_taxonomy_table, phylo_tree =tmp_phylo_tree)tmp_dataset$tidy_dataset()print(tmp_dataset)tmp_dataset$tax_table%<>%base::subset(Kingdom=="k__Archaea"|Kingdom=="k__Bacteria")print(tmp_dataset)tmp_dataset$filter_pollution(taxa =c("mitochondria", "chloroplast"))print(tmp_dataset)tmp_dataset$tidy_dataset()print(tmp_dataset)tmp_name<-purrr::map_chr(i, ~paste0(., "_me"))assign(tmp_name, tmp_dataset)rm(list =ls(pattern ="tmp_"))}## 5) Now, we calculate the taxa abundance at each taxonomic rank using ## `cal_abund()`. This function return a list called `taxa_abund` containing ## several data frame of the abundance information at each taxonomic rank. ## The list is stored in the microtable object automatically. ## It’s worth noting that the `cal_abund()` function can be used to solve ## some complex cases, such as supporting both the relative and absolute ## abundance calculation and selecting the partial taxonomic columns. More ## information can be found in the description of the ## [file2meco package](https://github.com/ChiLiubio/file2meco). ## In the same loop we also create a `trans_abund` class, which is used to ## transform taxonomic abundance data for plotting.for(iinssu18_data_sets){tmp_me<-get(purrr::map_chr(i, ~paste0(., "_me")))tmp_me$cal_abund()tmp_me_abund<-trans_abund$new(dataset =tmp_me, taxrank ="Phylum", ntaxa =12)tmp_me_abund$abund_data$Abundance<-tmp_me_abund$abund_data$Abundance/100tmp_me_abund_gr<-trans_abund$new( dataset =tmp_me, taxrank ="Phylum", ntaxa =12, groupmean ="TEMP")tmp_me_abund_gr$abund_data$Abundance<-tmp_me_abund_gr$abund_data$Abundance/100tmp_name<-purrr::map_chr(i, ~paste0(., "_me_abund"))assign(tmp_name, tmp_me_abund)tmp_name_gr<-purrr::map_chr(i, ~paste0(., "_me_abund_group"))assign(tmp_name_gr, tmp_me_abund_gr)rm(list =ls(pattern ="tmp_"))}## 6) I prefer to specify the order of taxa in these kinds of plots. ## We can look the top `ntaxa` (defined above) by accessing the ## `data_taxanames` character vector of each microtable object.## Now we can define the order. Once we do that, we will override the ## `data_taxanames` character vectors with the reordered vectors.ssu18_ps_work_tax_ord<-c("Alphaproteobacteria", "Gammaproteobacteria", "Acidobacteriota", "Actinobacteriota", "Bacteroidota", "Firmicutes", "Myxococcota", "Verrucomicrobiota", "Chloroflexi", "Planctomycetota", "Methylomirabilota", "Crenarchaeota")## 7) And one more little step before plotting. ## Here we **a**) specify a custom color palette and ## **b**) specify the sample order. top_level<-"Phylum"swel_col<-c("#2271B2", "#71B222", "#B22271")ssu18_colvec.tax<-c("#00463C","#FFD5FD","#00A51C","#C80B2A","#00C7F9","#FFA035", "#ED0DFD","#0063E5","#5FFFDE","#C00B6F","#00A090","#FF95BA")ssu18_samp_order<-c("P02_D00_010_C0A", "P04_D00_010_C0B", "P06_D00_010_C0C", "P08_D00_010_C0D", "P10_D00_010_C0E", "P01_D00_010_W3A", "P03_D00_010_W3B", "P05_D00_010_W3C", "P07_D00_010_W3D", "P09_D00_010_W3E", "P01_D00_010_W8A", "P03_D00_010_W8B", "P05_D00_010_W8C", "P07_D00_010_W8D", "P09_D00_010_W8E")## 8) Now we can generate plots (in a loop) for each faceted data set.for(iinssu18_data_sets){tmp_abund<-get(purrr::map_chr(i, ~paste0(., "_me_abund")))tmp_tax_order<-get(purrr::map_chr(i, ~paste0(., "_tax_ord")))tmp_abund$data_taxanames<-tmp_tax_ordertmp_facet_plot<-tmp_abund$plot_bar( color_values =ssu18_colvec.tax, others_color ="#323232", facet ="TEMP", xtext_keep =TRUE, xtext_type_hor =FALSE, legend_text_italic =FALSE, xtext_size =6, facet_color ="#cccccc", order_x =ssu18_samp_order)tmp_facet_name<-purrr::map_chr(i, ~paste0(., "_facet_plot"))assign(tmp_facet_name, tmp_facet_plot)rm(list =ls(pattern ="tmp_"))}## Then add a little formatting to the faceted plots.set_to_plot<-c("ssu18_ps_work_facet_plot")for(iinset_to_plot){tmp_get<-get(i)tmp_get<-tmp_get+theme_cowplot()+guides(fill =guide_legend( title =top_level, reverse =FALSE, keywidth =0.7, keyheight =0.7))+ylab(NULL)+xlab("Sample")+theme( panel.grid.major =element_blank(), panel.grid.minor =element_blank(), panel.background =element_rect(fill ="transparent", colour =NA), plot.background =element_rect(fill ="transparent", colour =NA), panel.border =element_rect(fill =NA, color ="black"), legend.text =element_text(size =7), legend.title =element_text(size =10), legend.position ="right", axis.text.y =element_text(size =8), axis.text.x =element_text(size =6, angle =90), strip.text =element_text(size =8, angle =0), axis.title =element_text(size =10))+ylab(NULL)+scale_y_continuous()assign(i, tmp_get)rm(list =ls(pattern ="tmp_"))}## And now plots for the group-means sets. We can use the same ## taxa order since that should not have changed.set_to_plot<-c("ssu18_ps_work_group_plot")for(iinssu18_data_sets){tmp_abund<-get(purrr::map_chr(i, ~paste0(., "_me_abund_group")))tmp_tax_order<-get(purrr::map_chr(i, ~paste0(., "_tax_ord")))tmp_abund$data_taxanames<-tmp_tax_ordertmp_group_plot<-tmp_abund$plot_bar( color_values =ssu18_colvec.tax, others_color ="#323232", xtext_keep =TRUE, xtext_type_hor =TRUE, legend_text_italic =FALSE, xtext_size =10, facet_color ="#cccccc")tmp_group_name<-purrr::map_chr(i, ~paste0(., "_group_plot"))assign(tmp_group_name, tmp_group_plot)rm(list =ls(pattern ="tmp_"))}## Let's also add a little formatting to the groupmean plots.for(iinset_to_plot){tmp_get<-get(i)tmp_get<-tmp_get+theme_cowplot()+ylab("Relative Abundance (% total reads)")+xlab("Temperature")+theme( panel.grid.major =element_blank(), panel.grid.minor =element_blank(), panel.background =element_rect(fill ="transparent", colour =NA), plot.background =element_rect(fill ="transparent", colour =NA), panel.border =element_rect(fill =NA, color ="black"), legend.position ="none", axis.text =element_text(size =8), axis.title =element_text(size =10))+scale_y_continuous()assign(i, tmp_get)rm(list =ls(pattern ="tmp_"))}## 10) Finally we use the `patchwork` package to combine the ## two plots and customize the look.## single index that acts as an index for referencing elements (variables) in a list## solution modified from this SO answerhttps://stackoverflow.com/a/54451460var_list<-list(var1 =ssu18_data_sets, var2 =c("FULL"))for(jin1:length(var_list$var1)){tmp_plot_final_name<-purrr::map_chr(var_list$var1[j], ~paste0(., "_", top_level, "_plot_final"))tmp_set_type<-var_list$var2[j]tmp_p_plot<-get(purrr::map_chr(var_list$var1[j], ~paste0(., "_group_plot")))tmp_m_plot<-get(purrr::map_chr(var_list$var1[j], ~paste0(., "_facet_plot")))tmp_plot_final<-tmp_p_plot+tmp_m_plottmp_plot_final<-tmp_plot_final+plot_annotation(tag_levels ="a")+plot_layout(widths =c(1, 2))&theme( plot.title =element_text(size =13), plot.subtitle =element_text(size =10), plot.tag =element_text(size =12), axis.title =element_text(size =10), axis.text =element_text(size =8))assign(tmp_plot_final_name, tmp_plot_final)rm(list =ls(pattern ="tmp_"))}for(iinssu18_data_sets){tmp_plot_final<-get(purrr::map_chr(i, ~paste0(., "_", top_level, "_plot_final")))ggplot2::ggsave(tmp_plot_final, file =paste0("include/pub/SOM/", i, "_tax_div_bar_plots.png", sep =""), height =2544, width =5043, units ='px', bg ="white", dpi =600)ggplot2::ggsave(tmp_plot_final, file =paste0("include/pub/SOM/", i, "_tax_div_bar_plots.pdf", sep =""), height =2544, width =5043, units ='px', bg ="white", dpi =600)rm(list =ls(pattern ="tmp_"))}file.rename("include/pub/SOM/ssu18_ps_work_tax_div_bar_plots.png", "include/pub/SOM/Supplementary_Figure_1.png")file.rename("include/pub/SOM/ssu18_ps_work_tax_div_bar_plots.pdf", "include/pub/SOM/Supplementary_Figure_1.pdf")
Supplementary Figure 2
Modifications
Post processing performed in Inkscape. Modifications include sample and variable renaming, and small adjustments in bar height/width.
its18_ps_work<-readRDS("include/pub/SOM/Supplementary_Figure_2.rds")swel_col<-c("#2271B2", "#71B222", "#B22271")its18_data_sets<-c("its18_ps_work")## 1) Choose the **number** of taxa to display and the ## taxonomic **level**. Aggregate the rest into "Other".top_hits<-14top_level<-"Order"## As above, in order to use `microeco`, we need to add the rank ## designation as a prefix to each taxa. For example, `Basidiomycota` ## is changed to `p__Basidiomycota`. for(iinits18_data_sets){tmp_get<-get(i)tmp_sam_data<-sample_data(tmp_get)tmp_tax_data<-data.frame(tax_table(tmp_get))tmp_tax_data$ASV_ID<-NULL# Some have, some do nottmp_tax_data$ASV_SEQ<-NULLtmp_tax_data$Kingdom<-paste("k__", tmp_tax_data$Kingdom, sep ="")tmp_tax_data$Phylum<-paste("p__", tmp_tax_data$Phylum, sep ="")tmp_tax_data$Class<-paste("c__", tmp_tax_data$Class, sep ="")tmp_tax_data$Order<-paste("o__", tmp_tax_data$Order, sep ="")tmp_tax_data$Family<-paste("f__", tmp_tax_data$Family, sep ="")tmp_tax_data$Genus<-paste("g__", tmp_tax_data$Genus, sep ="")tmp_tax_data<-as.matrix(tmp_tax_data)tmp_ps<-phyloseq(otu_table(tmp_get),tax_table(tmp_tax_data),tmp_sam_data)assign(i, tmp_ps)rm(list =ls(pattern ="tmp_"))}for(iinits18_data_sets){tmp_get<-get(i)tmp_otu_table<-data.frame(t(otu_table(tmp_get)))tmp_sample_info<-data.frame(sample_data(tmp_get))tmp_taxonomy_table<-data.frame(tax_table(tmp_get))tmp_dataset<-microtable$new(sample_table =tmp_sample_info, otu_table =tmp_otu_table, tax_table =tmp_taxonomy_table)tmp_dataset$tidy_dataset()print(tmp_dataset)tmp_dataset$tax_table%<>%base::subset(Kingdom=="k__Fungi")print(tmp_dataset)tmp_dataset$tidy_dataset()print(tmp_dataset)tmp_name<-purrr::map_chr(i, ~paste0(., "_me"))assign(tmp_name, tmp_dataset)rm(list =ls(pattern ="tmp_"))}## 5) Now, we calculate the taxa abundance at each taxonomic ## rank using `cal_abund()`. This function return a list called ## `taxa_abund` containing several data frame of the abundance ## information at each taxonomic rank. The list is stored in the ## microtable object automatically. It’s worth noting that the ## `cal_abund()` function can be used to solve some complex cases, ## such as supporting both the relative and absolute abundance ## calculation and selecting the partial taxonomic columns. ## More information can be found in the description of the ## [file2meco package](https://github.com/ChiLiubio/file2meco). ## In the same loop we also create a `trans_abund` class, which ## is used to transform taxonomic abundance data for plotting.for(iinits18_data_sets){tmp_me<-get(purrr::map_chr(i, ~paste0(., "_me")))tmp_me$cal_abund()tmp_me_abund<-trans_abund$new(dataset =tmp_me, taxrank =top_level, ntaxa =top_hits)tmp_me_abund$abund_data$Abundance<-tmp_me_abund$abund_data$Abundance/100tmp_me_abund_gr<-trans_abund$new( dataset =tmp_me, taxrank =top_level, ntaxa =top_hits, groupmean ="TEMP")tmp_me_abund_gr$abund_data$Abundance<-tmp_me_abund_gr$abund_data$Abundance/100tmp_name<-purrr::map_chr(i, ~paste0(., "_me_abund"))assign(tmp_name, tmp_me_abund)tmp_name_gr<-purrr::map_chr(i, ~paste0(., "_me_abund_group"))assign(tmp_name_gr, tmp_me_abund_gr)rm(list =ls(pattern ="tmp_"))}## 6) I prefer to specify the order of taxa in these kinds of plots. ## We can look the top `ntaxa` (defined above) by accessing the ## `data_taxanames` character vector of each microtable object.its18_ps_work_tax_ord<-rev(c("k_Fungi", "p_Ascomycota", "c_Agaricomycetes", "Agaricales", "Archaeorhizomycetales", "Capnodiales", "Eurotiales", "Geastrales", "Glomerales", "Helotiales", "Hypocreales", "Saccharomycetales", "Trichosporonales", "Xylariales"))## And one more little step before plotting. Here we ## **a**) specify a custom color palette and ## **b**) specify the sample order. its18_colvec.tax<-rev(c("#323232", "#004949", "#924900", "#490092", "#6db6ff", "#920000", "#ffb6db", "#24ff24", "#006ddb", "#db6d00", "#b66dff", "#ffff6d", "#009292", "#b6dbff", "#ff6db6"))its18_samp_order<-c("P02_D00_010_C0A", "P04_D00_010_C0B", "P06_D00_010_C0C", "P08_D00_010_C0D", "P10_D00_010_C0E", "P01_D00_010_W3A", "P03_D00_010_W3B", "P07_D00_010_W3D", "P09_D00_010_W3E", "P01_D00_010_W8A", "P03_D00_010_W8B", "P05_D00_010_W8C", "P07_D00_010_W8D")## 8) Now we can generate plots (in a loop) for each faceted data set.for(iinits18_data_sets){tmp_abund<-get(purrr::map_chr(i, ~paste0(., "_me_abund")))tmp_tax_order<-get(purrr::map_chr(i, ~paste0(., "_tax_ord")))tmp_abund$data_taxanames<-tmp_tax_ordertmp_facet_plot<-tmp_abund$plot_bar( color_values =its18_colvec.tax, others_color ="#323232", facet ="TEMP", xtext_keep =TRUE, xtext_type_hor =FALSE, legend_text_italic =FALSE, xtext_size =6, facet_color ="#cccccc", order_x =its18_samp_order)tmp_facet_name<-purrr::map_chr(i, ~paste0(., "_facet_plot"))assign(tmp_facet_name, tmp_facet_plot)rm(list =ls(pattern ="tmp_"))}## Then add a little formatting to the faceted plots.set_to_plot<-c("its18_ps_work_facet_plot")for(iinset_to_plot){tmp_get<-get(i)tmp_get<-tmp_get+theme_cowplot()+guides(fill =guide_legend( title =top_level, reverse =FALSE, keywidth =0.7, keyheight =0.7))+ylab(NULL)+xlab("Sample")+theme( panel.grid.major =element_blank(), panel.grid.minor =element_blank(), panel.background =element_rect(fill ="transparent", colour =NA), plot.background =element_rect(fill ="transparent", colour =NA), panel.border =element_rect(fill =NA, color ="black"), legend.text =element_text(size =7), legend.title =element_text(size =10), legend.position ="right", axis.text.y =element_text(size =8), axis.text.x =element_text(size =6, angle =90), strip.text =element_text(size =8, angle =0), axis.title =element_text(size =10))+ylab(NULL)+scale_y_continuous()assign(i, tmp_get)rm(list =ls(pattern ="tmp_"))}## And now plots for the groupmeans sets. ## We can use the same taxa order since that ## should not have changed.set_to_plot<-c("its18_ps_work_group_plot")for(iinits18_data_sets){tmp_abund<-get(purrr::map_chr(i, ~paste0(., "_me_abund_group")))tmp_tax_order<-get(purrr::map_chr(i, ~paste0(., "_tax_ord")))tmp_abund$data_taxanames<-tmp_tax_ordertmp_group_plot<-tmp_abund$plot_bar( color_values =its18_colvec.tax, others_color ="#323232", xtext_keep =TRUE, xtext_type_hor =TRUE, legend_text_italic =FALSE, xtext_size =10, facet_color ="#cccccc")tmp_group_name<-purrr::map_chr(i, ~paste0(., "_group_plot"))assign(tmp_group_name, tmp_group_plot)rm(list =ls(pattern ="tmp_"))}## Let's also add a little formatting to the groupmean plots.for(iinset_to_plot){tmp_get<-get(i)tmp_get<-tmp_get+theme_cowplot()+ylab("Relative Abundance (% total reads)")+xlab("Temperature")+theme( panel.grid.major =element_blank(), panel.grid.minor =element_blank(), panel.background =element_rect(fill ="transparent", colour =NA), plot.background =element_rect(fill ="transparent", colour =NA), panel.border =element_rect(fill =NA, color ="black"), legend.position ="none", axis.text =element_text(size =8), axis.title =element_text(size =10))+scale_y_continuous()assign(i, tmp_get)rm(list =ls(pattern ="tmp_"))}## 10) Finally we use the `patchwork` package to combine the ## two plots and customize the look.## single index that acts as an index for referencing elements (variables) in a list## solution modified from this SO answerhttps://stackoverflow.com/a/54451460var_list<-list(var1 =its18_data_sets, var2 =c("FULL"))for(jin1:length(var_list$var1)){tmp_plot_final_name<-purrr::map_chr(var_list$var1[j], ~paste0(., "_", top_level, "_plot_final"))tmp_set_type<-var_list$var2[j]tmp_p_plot<-get(purrr::map_chr(var_list$var1[j], ~paste0(., "_group_plot")))tmp_m_plot<-get(purrr::map_chr(var_list$var1[j], ~paste0(., "_facet_plot")))tmp_plot_final<-tmp_p_plot+tmp_m_plottmp_plot_final<-tmp_plot_final+plot_annotation(tag_levels ="a")+plot_layout(widths =c(1, 2))&theme( plot.title =element_text(size =13), plot.subtitle =element_text(size =10), plot.tag =element_text(size =12), axis.title =element_text(size =10), axis.text =element_text(size =8))assign(tmp_plot_final_name, tmp_plot_final)rm(list =ls(pattern ="tmp_"))}for(iinits18_data_sets){tmp_plot_final<-get(purrr::map_chr(i, ~paste0(., "_", top_level, "_plot_final")))ggplot2::ggsave(tmp_plot_final, file =paste0("include/pub/SOM/", i, "_tax_div_bar_plots.png", sep =""), height =2544, width =5043, units ='px', bg ="white", dpi =600)ggplot2::ggsave(tmp_plot_final, file =paste0("include/pub/SOM/", i, "_tax_div_bar_plots.pdf", sep =""), height =2544, width =5043, units ='px', bg ="white", dpi =600)rm(list =ls(pattern ="tmp_"))}file.rename("include/pub/SOM/its18_ps_work_tax_div_bar_plots.png", "include/pub/SOM/Supplementary_Figure_2.png")file.rename("include/pub/SOM/its18_ps_work_tax_div_bar_plots.pdf", "include/pub/SOM/Supplementary_Figure_2.pdf")
Supplementary Figure 3
Modifications
Post processing performed in Inkscape. Modifications include sample and variable renaming, removing non-significant values from plots, and changing significant p-values to asterisks (*).
remove(list =ls())load("include/pub/SOM/Supplementary_Figure_3.rdata")swel_col<-c("#2271B2", "#71B222", "#B22271")div_test_plot_jjs<-function(divtest, chart, colour, posthoc, threshold){if(missing(chart)){chart="box"}if(missing(posthoc)){posthoc=FALSE}if((names(divtest)[1]!="data")&(names(divtest)[2]!="normality.pvalue"))stop("The input object does not seem to be a div_test output.")divtestdata<-divtest$datadivtestdata$Group<-as.factor(divtestdata$Group)divtestdata$Group<-factor(divtestdata$Group, levels =as.character(unique(divtestdata$Group)))if(missing(colour)||(length(colour)<divtest$groups)){getPalette<-colorRampPalette(brewer.pal(divtest$groups, "Paired"))colour<-getPalette(divtest$groups)}if(posthoc==TRUE){if(is.na(names(divtest)[7]))stop("The input div_test object does not seem to contain pairwise posthoc data. Re-run div_test() using 'posthoc=TRUE' argument.")if(divtest[7]=="Tukey post-hoc test"){combinations<-matrix(gsub(" $", "", gsub("^ ", "", unlist(strsplit(as.character(rownames(divtest$posthoc)), "-", fixed =TRUE)))), ncol =2, byrow =TRUE)pvalue<-round(divtest$posthoc[, 4], 4)pairwisetable<-as.data.frame(cbind(combinations, pvalue))colnames(pairwisetable)<-c("group1", "group2", "p")}if(divtest[7]=="Dunn test with Benjamini-Hochberg correction"){combinations<-matrix(gsub(" $", "", gsub("^ ", "", unlist(strsplit(as.character(rownames(divtest$posthoc)), "-", fixed =TRUE)))), ncol =2, byrow =TRUE)pvalue<-round(divtest$posthoc[, 3], 4)pairwisetable<-as.data.frame(cbind(combinations, pvalue))colnames(pairwisetable)<-c("group1", "group2", "p")}pairwisetable[, 1]<-as.character(pairwisetable[, 1])pairwisetable[, 2]<-as.character(pairwisetable[, 2])pairwisetable[, 3]<-as.numeric(as.character(pairwisetable[, 3]))if(!missing(threshold)){pairwisetable<-pairwisetable[which(pairwisetable$p<threshold), ]}sortedgroups<-unique(sort(c(pairwisetable$group1, pairwisetable$group2)))datamax<-round(max(divtest$data[which(divtest$data$Group%in%sortedgroups), 3]))datamin<-round(min(divtest$data[which(divtest$data$Group%in%sortedgroups), 3]))datarange<-datamax-dataminby<-datarange*0.1min<-datamaxmax<-min+(by*nrow(pairwisetable))ypos<-seq(min, max, by)[-1]pairwisetable$ypos<-ypos}if(chart=="box"){plot<-ggboxplot(divtestdata, x ="Group", y ="Value", outlier.size =3, color ="Group", fill ="Group", x.text.angle =0)+ylab("Effective no. of Taxon Units")+xlab("Treatment")+#scale_colour_manual(values = scales::alpha(colour, 1)) + scale_colour_manual(values=c("#191919", "#191919", "#191919"))+scale_fill_manual(values =scales::alpha(colour, 1))+scale_linetype_manual()if(posthoc==TRUE){plot<-suppressWarnings(plot+stat_pvalue_manual(pairwisetable, label ="p", y.position ="ypos"))}return(plot)}if(chart=="jitter"){plot<-ggboxplot(divtestdata, x ="Group", y ="Value", color ="Group", add ="jitter", width =0, x.text.angle =45)+ylab("Effective no. of Taxon Units")+xlab("Treatment")+scale_colour_manual(values =scales::alpha(colour, 0))if(posthoc==TRUE){plot<-suppressWarnings(plot+stat_pvalue_manual(pairwisetable, label ="p", y.position ="ypos"))}print(plot)}if(chart=="violin"){plot<-ggviolin(divtestdata, x ="Group", y ="Value", color ="Group", fill ="Group", x.text.angle =45)+ylab("Effective no. of Taxon Units")+xlab("Treatment")+scale_fill_manual(values =scales::alpha(colour, 0.1))+scale_colour_manual(values =scales::alpha(colour, 1))if(posthoc==TRUE){plot<-suppressWarnings(plot+stat_pvalue_manual(pairwisetable, label ="p", y.position ="ypos"))}print(plot)}}rm(list =ls(pattern ="_adt_plot"))for(iinobjects(pattern ="_adt")){tmp_name<-purrr::map_chr(i, ~paste0(., "_plot"))tmp_get<-get(i)tmp_df<-div_test_plot_jjs(tmp_get, chart ="box", colour =swel_col, posthoc =TRUE)tmp_df<-ggpar(tmp_df, legend ="none")print(tmp_name)assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_"))}for(iinobjects(pattern ="_adt_plot")){tmp_split<-stringr::str_split(i, "_")if(tmp_split[[1]][3]=="work"){tmp_ds<-"f"tmp_name1<-"FULL"}elseif(tmp_split[[1]][3]=="filt"){tmp_ds<-"l"tmp_name1<-"FILT"}elseif(tmp_split[[1]][3]=="perfect"){tmp_ds<-"r"tmp_name1<-"PERfect"}elseif(tmp_split[[1]][3]=="pime"){tmp_ds<-"p"tmp_name1<-"PIME"}if(tmp_split[[1]][4]=="q0"){tmp_hill<-"0"tmp_name2<-"Observed"}elseif(tmp_split[[1]][4]=="q1"){tmp_hill<-"1"tmp_name2<-"Shannon exponential"}elseif(tmp_split[[1]][4]=="q2"){tmp_hill<-"2"tmp_name2<-"Inverse Simpson"}tmp_var<-paste(tmp_split[[1]][1], "_asv", tmp_ds, tmp_hill, "_lab", sep ="")tmp_name<-paste(tmp_name1, " (", tmp_name2, ")", sep ="")assign(tmp_var, tmp_name)rm(list =ls(pattern ="tmp_"))}ssu18_ps_work_q0_adt_plot<-ssu18_ps_work_q0_adt_plot+theme(axis.title.x =element_blank())+ggtitle(ssu18_asvf0_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_filt_q0_adt_plot<-ssu18_ps_filt_q0_adt_plot+theme(axis.title.y =element_blank(), axis.title.x =element_blank())+ggtitle(ssu18_asvl0_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_perfect_q0_adt_plot<-ssu18_ps_perfect_q0_adt_plot+theme(axis.title.y =element_blank(), axis.title.x =element_blank())+ggtitle(ssu18_asvr0_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_pime_q0_adt_plot<-ssu18_ps_pime_q0_adt_plot+theme(axis.title.y =element_blank(), axis.title.x =element_blank())+ggtitle(ssu18_asvp0_lab)+theme(plot.title =element_text(size =20, face ="bold"))#####################ssu18_ps_work_q1_adt_plot<-ssu18_ps_work_q1_adt_plot+theme(axis.title.x =element_blank())+ggtitle(ssu18_asvf1_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_filt_q1_adt_plot<-ssu18_ps_filt_q1_adt_plot+theme(axis.title.y =element_blank(), axis.title.x =element_blank())+ggtitle(ssu18_asvl1_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_perfect_q1_adt_plot<-ssu18_ps_perfect_q1_adt_plot+theme(axis.title.y =element_blank(), axis.title.x =element_blank())+ggtitle(ssu18_asvr1_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_pime_q1_adt_plot<-ssu18_ps_pime_q1_adt_plot+theme(axis.title.y =element_blank(), axis.title.x =element_blank())+ggtitle(ssu18_asvp1_lab)+theme(plot.title =element_text(size =20, face ="bold"))#####################ssu18_ps_work_q2_adt_plot<-ssu18_ps_work_q2_adt_plot+ggtitle(ssu18_asvf2_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_filt_q2_adt_plot<-ssu18_ps_filt_q2_adt_plot+theme(axis.title.y =element_blank())+ggtitle(ssu18_asvl2_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_perfect_q2_adt_plot<-ssu18_ps_perfect_q2_adt_plot+theme(axis.title.y =element_blank())+ggtitle(ssu18_asvr2_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_pime_q2_adt_plot<-ssu18_ps_pime_q2_adt_plot+theme(axis.title.y =element_blank())+ggtitle(ssu18_asvp2_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_alph_div_plots_asv<-ggarrange(ssu18_ps_work_q0_adt_plot,ssu18_ps_filt_q0_adt_plot,ssu18_ps_perfect_q0_adt_plot,ssu18_ps_pime_q0_adt_plot,ssu18_ps_work_q1_adt_plot,ssu18_ps_filt_q1_adt_plot,ssu18_ps_perfect_q1_adt_plot,ssu18_ps_pime_q1_adt_plot,ssu18_ps_work_q2_adt_plot,ssu18_ps_filt_q2_adt_plot,ssu18_ps_perfect_q2_adt_plot,ssu18_ps_pime_q2_adt_plot, ncol =4, nrow =3)ggplot2::ggsave(ssu18_alph_div_plots_asv, file ="include/pub/SOM/ssu18_alph_div_plots_asv.png", height =5852, width =7449, units ='px', bg ="white", dpi =600)ggplot2::ggsave(ssu18_alph_div_plots_asv, file ="include/pub/SOM/ssu18_alph_div_plots_asv.pdf", height =5852, width =7449, units ='px', bg ="white", dpi =600)file.rename("include/pub/SOM/ssu18_alph_div_plots_asv.png","include/pub/SOM/Supplementary_Figure_3.png")file.rename("include/pub/SOM/ssu18_alph_div_plots_asv.pdf","include/pub/SOM/Supplementary_Figure_3.pdf")
Supplementary Figure 4
Modifications
Post processing performed in Inkscape. Modifications include sample and variable renaming, removing non-significant values from plots, and changing significant p-values to asterisks (*).