This workflow contains diversity assessments for the 2018 high temperature data sets. In order to run the workflow, you either need to first run the DADA2 Workflowand the Data Preparation workflowor download the files linked below. See the Data Availability page for complete details.
Workflow Input
Files needed to run this workflow can be downloaded from figshare.
16s rRNA
To account for presence of rare sequence variants caused by sequencing errors or other technical artifacts, we use Hill numbers (Alberdi and Gilbert 2019a). Hill numbers allow the weight put on rare versus abundant sequence variants to be scaled while providing intuitive comparisons of diversity levels using “effective number of ASVs” as a measuring unit. This approach allows for balancing the over representation of rare ASVs that might be inflated due to sequencing errors.
We will then use Shapiro-Wilk tests to test for normalcy and then, depending on the results, either use parametric ANOVA or non-parametric Kruskal-Wallis to compare alpha diversity among treatments.
Calculate Hill Numbers
To calculate Hill numbers, we use the R package hilldiv(Alberdi and Gilbert 2019b). We calculate three metrics that put more or less weight on common species:
Observed richness, where q-value = 0.
Shannon exponential, which weighs ASVs by their frequency, where q-value = 1.
Simpson multiplicative inverse, which over weighs abundant ASVs, where q-value = 2.
We perform each analysis against the Full (unfiltered) data set as well as the Arbitrary, PERfect and PIME, filtered data sets using the function hill_div.
The command is as follows:
hill_div(count = x, qvalue = i, tree = ultrametric_tree)
where x is the sample by ASV table, i is the q-value corresponding to the metric of interest and tree is an ultrametric formatted phylogenetic tree if you choose to look at lineage, rather than ASV, diversity.
We first transform all the data to relative abundance values, and compute new trees.
Now we summarize the data for each sample against all three metrics. The table contains the results of ASV diversity estimates from the full data set and the three filtered data sets.
The suffix _fi indicates metrics for the Arbitrary data set. The suffix _pe indicates metrics for the PERfect data set and the suffix _pet indicates the lineage diversity for the PERfect data set.
The suffix _pi indicates metrics for the PIME data set and the suffix _pit indicates the lineage diversity for the PIME data set.
Hill Summary
(16S rRNA) Table 1 | Hill numbers for samples in the unfiltered & filtered data sets.
Normality Tests
Before running significance tests, we need to know if data is normally distributed, which will tell use whether to use a parametric or non-parametric test. To test if the data are normally distributed, we use the Shapiro-Wilk Normality test and the Bartlett Test of Homogeneity of Variances.
If the p-values are both not significant (p > 0.05) from the tests, we accept the null hypothesis (that the results are normally distributed) and test for significance between samples using an ANOVA. If the p-values are both significant (p < 0.05), we reject the null hypothesis (that the results are normally distributed) and test for significance between samples using Kruskal-Wallis (non-parametric equivalent of ANOVA).
The commands are as follows:
shapiro.test(x), where x is a numeric vector of alpha diversity values from the sample data table.
bartlett.test(Value ~ Group, data = df) Where Value is the metric of interest, Group in the treatment to compare, and df is the data frame.
And then the Bartlett Test of Homogeneity of Variances.
Show the code
ssu18_div_tab_asv<-ssu_tab_alpha_divssu18_bart_tests_asv<-c()for(iincolnames(ssu18_div_tab_asv[,7:ncol(ssu18_div_tab_asv)])){tmp_name<-purrr::map_chr(i, ~paste0("ssu18_bart_asv_", .))ssu18_bart_tests_asv<-append(ssu18_bart_tests_asv, tmp_name)tmp_test<-eval(bartlett.test(ssu18_div_tab_asv[[i]]~TEMP, data =ssu18_div_tab_asv))tmp_test$data.name<-tmp_nameassign(tmp_name, tmp_test)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bart_")
Here we see which Shapiro-Wilk Normality and Bartlett tests were significant and which were not. So wherever the value of both p-values in > 0.05 we can use an ANOVA, otherwise we use Kruskal-Wallis.
(16S rRNA) Table 2 | Summary of Normality Tests for unfiltered & filtered data sets. P-values in red indicate significance (p-value < 0.05).
Significance Tests
To begin, we need to create a hierarchy variable; a two-column matrix specifying the relationship between samples (first column) and groups (second column).
Again, we start by testing significance of ASV diversity for the data sets against each of the three metrics using the div_test function.
The command is as follows:
div_test(countable = x, qvalue = i, hierarchy = hier,
tree = ultrametric_tree, posthoc = TRUE)
where x is ASV by sample table, i is the q-value corresponding to the metric of interest, hier is the hierarchy matrix, tree is an ultrametric formatted phylogenetic tree (if you choose to explore lineage diversity), and posthoc indicates whether to run post hoc pairwise analyses.
(16S rRNA) Table 3 | Summary of significance tests for unfiltered & filtered data sets. P-values in red for post hoc analysis indicate significance (p-value < 0.05).
PostHoc Analyses
First let’s check the results of each posthoc analysis.
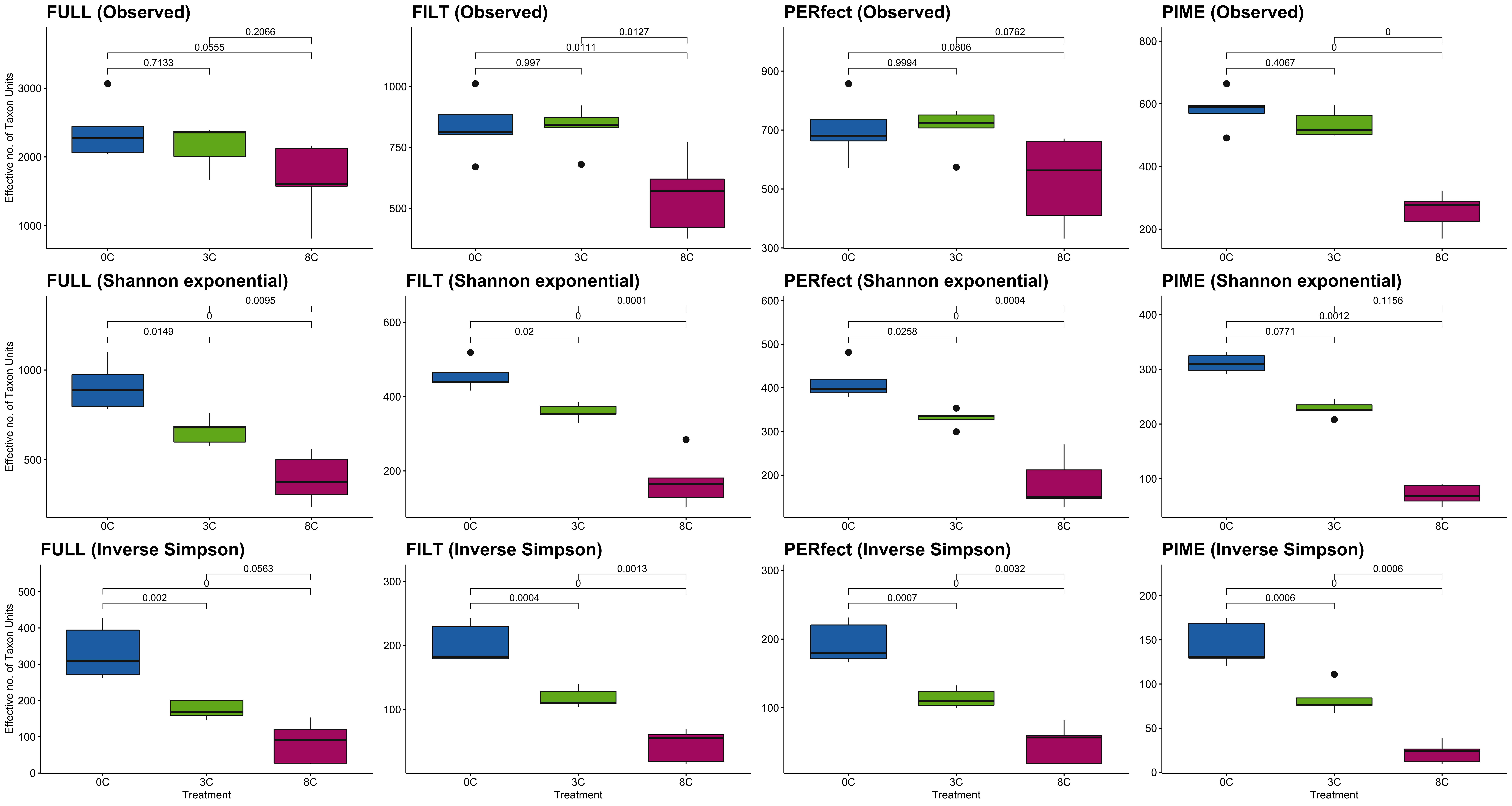
Now we can plot the results from the posthoc analyses for each metric and data set using the function div_test_plot_jjs. I modified the original function (div_test_plot) to control a little of the formatting.
where x is the results from the div_test function, "type" is chart type (box, jitter, or violin), colour is is a color palette, posthoc indicates whether to run posthoc pairwise analyses, and value is the maximum p-value to show in pairwise posthoc results. WARNING if none of the posthoc results are below the specified threshold, the function will throw an error. Therefore, until this is fixed, all posthoc values are shown.
Posthoc adjusted p-values given for each pairwise comparison.
(16S rRNA) Figure 1 | Alpha diversity plots. Top row = Observed; middle row = Shannon exponential; bottom row = Inverse Simpson. From left to right, Full (unfiltered) data, Arbitrary filtered, PERfect filtered, PIME filtered.
ITS
To account for presence of rare sequence variants caused by sequencing errors or other technical artifacts, we use Hill numbers (Alberdi and Gilbert 2019a). Hill numbers allow the weight put on rare versus abundant sequence variants to be scaled while providing intuitive comparisons of diversity levels using “effective number of ASVs” as a measuring unit. This approach allows for balancing the over representation of rare ASVs that might be inflated due to sequencing errors.
We will then use Shapiro-Wilk tests to test for normalcy and then, depending on the results, either use parametric ANOVA or non-parametric Kruskal-Wallis to compare alpha diversity among treatments.
Calculate Hill Numbers
To calculate Hill numbers, we use the R package hilldiv(Alberdi and Gilbert 2019b). We calculate three metrics that put more or less weight on common species:
Observed richness, where q-value = 0.
Shannon exponential, which weighs ASVs by their frequency, where q-value = 1.
Simpson multiplicative inverse, which over weighs abundant ASVs, where q-value = 2.
We perform each analysis against the Full (unfiltered) data set as well as the Arbitrary, PERfect and PIME, filtered data sets using the function hill_div.
The command is as follows:
hill_div(count = x, qvalue = i, tree = ultrametric_tree)
where x is the sample by ASV table, i is the q-value corresponding to the metric of interest and tree is an ultrametric formatted phylogenetic tree, however this really doesn’t apply for ITS data anyway.
We first transform all the data to relative abundance values, and compute new trees.
Now we summarize the data for each sample against all three metrics. The table contains the results of ASV diversity estimates from the full data set and the three filtered data sets.
The suffix _fi indicates metrics for the Arbitrary data set. The suffix _pe indicates metrics for the PERfect data set and the suffix _pet indicates the lineage diversity for the PERfect data set.
The suffix _pi indicates metrics for the PIME data set and the suffix _pit indicates the lineage diversity for the PIME data set.
Hill Summary
(ITS) Table 1 | Hill numbers for samples in the unfiltered & filtered data sets.
Normality Tests
Before running significance tests, we need to know if data is normally distributed, which will tell use whether to use a parametric or non-parametric test. To test if the data are normally distributed, we use the Shapiro-Wilk Normality test and the Bartlett Test of Homogeneity of Variances.
If the p-values are both not significant (p > 0.05) from the tests, we accept the null hypothesis (that the results are normally distributed) and test for significance between samples using an ANOVA. If the p-values are both significant (p < 0.05), we reject the null hypothesis (that the results are normally distributed) and test for significance between samples using Kruskal-Wallis (non-parametric equivalent of ANOVA).
The commands are as follows:
shapiro.test(x), where x is a numeric vector of alpha diversity values from the sample data table.
bartlett.test(Value ~ Group, data = df) Where Value is the metric of interest, Group in the treatment to compare, and df is the data frame.
And then the Bartlett Test of Homogeneity of Variances.
Show the code
its18_div_tab_asv<-its_tab_alpha_divits18_bart_tests_asv<-c()for(iincolnames(its18_div_tab_asv[,7:ncol(its18_div_tab_asv)])){tmp_name<-purrr::map_chr(i, ~paste0("its18_bart_asv_", .))its18_bart_tests_asv<-append(its18_bart_tests_asv, tmp_name)tmp_test<-eval(bartlett.test(its18_div_tab_asv[[i]]~TEMP, data =its18_div_tab_asv))tmp_test$data.name<-tmp_nameassign(tmp_name, tmp_test)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bart_")
Here we see which Shapiro-Wilk Normality and Bartlett tests were significant and which were not. So wherever the value of both p-values in > 0.05 we can use an ANOVA, otherwise we use Kruskal-Wallis.
(ITS) Table 2 | Summary of Normality Tests for unfiltered & filtered data sets. P-values in red indicate significance (p-value < 0.05).
Significance Tests
To begin, we need to create a hierarchy variable; a two-column matrix specifying the relationship between samples (first column) and groups (second column).
Again, we start by testing significance of ASV diversity for the data sets against each of the three metrics using the div_test function.
The command is as follows:
div_test(countable = x, qvalue = i, hierarchy = hier,
tree = ultrametric_tree, posthoc = TRUE)
where x is ASV by sample table, i is the q-value corresponding to the metric of interest, hier is the hierarchy matrix, tree is an ultrametric formatted phylogenetic tree (if you choose to explore lineage diversity), and posthoc indicates whether to run post hoc pairwise analyses.
(ITS) Table 3 | Summary of significance tests for unfiltered & filtered data sets. P-values in red for post hoc analysis indicate significance (p-value < 0.05).
PostHoc Analyses
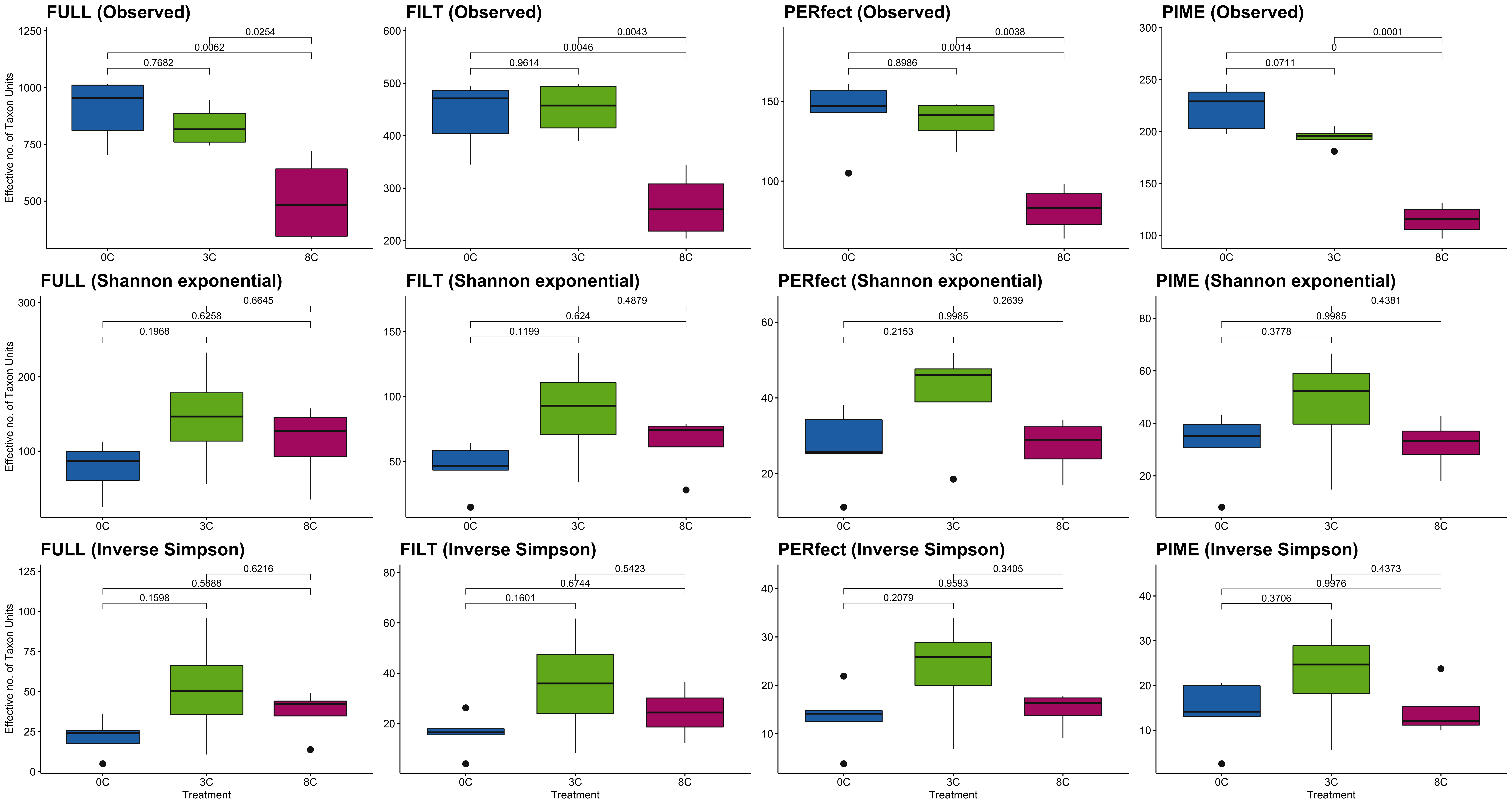
First let’s check the results of each posthoc analysis.
Now we can plot the results from the posthoc analyses for each metric and data set using the function div_test_plot_jjs. I modified the original function (div_test_plot) to control a little of the formatting.
where x is the results from the div_test function, "type" is chart type (box, jitter, or violin), colour is is a color palette, posthoc indicates whether to run posthoc pairwise analyses, and value is the maximum p-value to show in pairwise posthoc results. WARNING if none of the posthoc results are below the specified threshold, the function will throw an error. Therefore, until this is fixed, all posthoc values are shown.
The source code for this page can be accessed on GitHub by clicking this link.
Data Availability
Data generated in this workflow and the Rdata need to run the workflow can be accessed on figshare at 10.25573/data.16826779.
Last updated on
[1] "2025-08-23 12:57:03 PDT"
References
Alberdi, Antton, and M Thomas P Gilbert. 2019a. “A Guide to the Application of Hill Numbers to DNA-Based Diversity Analyses.”Molecular Ecology Resources 19 (4): 804–17. https://doi.org/10.1111/1755‐0998.13014.
———. 2019b. “Hilldiv: An r Package for the Integral Analysis of Diversity Based on Hill Numbers.”bioRxiv, 545665. https://doi.org/10.1101/545665.
Source Code
---title: "5. Alpha Diversity Estimates"description: | Reproducible workflow to assess alpha diversity across temperature treatments using Hill numbers.format: html: toc: true toc-depth: 3---```{r}#| message: false#| results: hide#| code-fold: true#| code-summary: "Click here for setup information"knitr::opts_chunk$set(echo =TRUE, eval =FALSE)set.seed(119)#library(conflicted)library(phyloseq); packageVersion("phyloseq")library(Biostrings); packageVersion("Biostrings")pacman::p_load(tidyverse, hilldiv, microbiome, phytools, phangorn, pairwiseAdonis, naniar, labdsv, patchwork, agricolae, ggpubr, reactable, downloadthis, captioner,install =FALSE, update =FALSE)source("hack_code/div_test_plot_jjs.R")source("hack_code/div_test_jjs.R")options(scipen=999)knitr::opts_current$get(c("cache","cache.path","cache.rebuild","dependson","autodep"))``````{r}#| include: false#| eval: true## Load to build page only #2remove(list =ls())load("page_build/alpha_wf.rdata")``````{r}#| include: false#| eval: true# Create the caption(s) with captionercaption_tab_ssu <-captioner(prefix ="(16S rRNA) Table", suffix =" |", style ="b")caption_fig_ssu <-captioner(prefix ="(16S rRNA) Figure", suffix =" |", style ="b")caption_tab_its <-captioner(prefix ="(ITS) Table", suffix =" |", style ="b")caption_fig_its <-captioner(prefix ="(ITS) Figure", suffix =" |", style ="b")# Create a function for referring to the tables in textref <-function(x) str_extract(x, "[^|]*") %>%trimws(which ="right", whitespace ="[ ]")``````{r}#| include: false#| eval: truesource(file.path("assets", "functions.R"))source("assets/captions/captions_alpha.R")```# SynopsisThis workflow contains diversity assessments for the 2018 high temperature data sets. In order to run the workflow, you either need to first run the [DADA2 Workflow](dada2.html) **and** the [Data Preparation workflow](data-prep.html) **or** download the files linked below. See the [Data Availability](data-availability.html) page for complete details.## Workflow InputFiles needed to run this workflow can be downloaded from figshare. <iframe src="https://widgets.figshare.com/articles/14690739/embed?show_title=1" width="100%" height="251" allowfullscreen frameborder="0"></iframe><iframe src="https://widgets.figshare.com/articles/14701440/embed?show_title=1" width="100%" height="351" allowfullscreen frameborder="0"></iframe># 16s rRNA```{r}#| include: false## Initial Load for ANALYSIS #1set.seed(119)## ASV FULL AND PIME AND PERfectssu18_ps_work <-readRDS("files/data-prep/rdata/ssu18_ps_work.rds")ssu18_ps_filt <-readRDS("files/filtering/arbitrary/rdata/ssu18_ps_filt.rds")ssu18_ps_perfect <-readRDS("files/filtering/perfect/rdata/ssu18_ps_perfect.rds")ssu18_ps_pime <-readRDS("files/filtering/pime/rdata/ssu18_ps_pime.rds")``````{r}#| echo: falseswel_col <-c("#2271B2", "#71B222", "#B22271")```To account for presence of rare sequence variants caused by sequencing errors or other technical artifacts, we use Hill numbers [@alberdi2019guide]. Hill numbers allow the weight put on rare versus abundant sequence variants to be scaled while providing intuitive comparisons of diversity levels using “effective number of ASVs” as a measuring unit. This approach allows for balancing the over representation of rare ASVs that might be inflated due to sequencing errors.We will then use Shapiro-Wilk tests to test for normalcy and then, depending on the results, either use parametric ANOVA or non-parametric Kruskal-Wallis to compare alpha diversity among treatments.## Calculate Hill NumbersTo calculate Hill numbers, we use the R package `hilldiv`[@alberdi2019hilldiv]. We calculate three metrics that put more or less weight on common species:1) Observed richness, where `q-value = 0`.2) Shannon exponential, which weighs ASVs by their frequency, where `q-value = 1`.3) Simpson multiplicative inverse, which over weighs abundant ASVs, where `q-value = 2`.We perform each analysis against the Full (unfiltered) data set as well as the Arbitrary, PERfect and PIME, filtered data sets using the function `hill_div`.The command is as follows:```hill_div(count = x, qvalue = i, tree = ultrametric_tree)```where `x` is the sample by ASV table, `i` is the q-value corresponding to the metric of interest and `tree` is an ultrametric formatted phylogenetic tree if you choose to look at lineage, rather than ASV, diversity.```{r}#| echo: false## this is for subsettig samples..rm(ssu18_asv_pime_sum)ssu18_alpha_ds <-c("ssu18_ps_work", "ssu18_ps_filt", "ssu18_ps_perfect", "ssu18_ps_pime")rm(list =ls(pattern ="_0"))rm(list =ls(pattern ="_3"))rm(list =ls(pattern ="_8"))for (i in ssu18_alpha_ds) { tmp_get <-get(i) tmp_0 <-prune_samples(c("P10_D00_010_C0E", "P02_D00_010_C0A", "P04_D00_010_C0B", "P06_D00_010_C0C", "P08_D00_010_C0D"), tmp_get) tmp_3 <-prune_samples(c("P01_D00_010_W3A", "P03_D00_010_W3B", "P05_D00_010_W3C", "P07_D00_010_W3D", "P09_D00_010_W3E"), tmp_get) tmp_8 <-prune_samples(c("P01_D00_010_W8A", "P03_D00_010_W8B", "P05_D00_010_W8C", "P07_D00_010_W8D", "P09_D00_010_W8E"), tmp_get) tmp_0 <-prune_taxa(taxa_sums(tmp_0) >0, tmp_0) tmp_3 <-prune_taxa(taxa_sums(tmp_3) >0, tmp_3) tmp_8 <-prune_taxa(taxa_sums(tmp_8) >0, tmp_8) tmp_ps_name_0 <- purrr::map_chr(i, ~paste0(., "_0"))assign(tmp_ps_name_0, tmp_0) tmp_ps_name_3 <- purrr::map_chr(i, ~paste0(., "_3"))assign(tmp_ps_name_3, tmp_3) tmp_ps_name_8 <- purrr::map_chr(i, ~paste0(., "_8"))assign(tmp_ps_name_8, tmp_8)rm(list =ls(pattern ="tmp_"))}tmp_objects <-data.frame(c("FULL data set (0C)", "FULL data set (3C)", "FULL data set (8C)", "Arbitrary filter data (0C)", "Arbitrary filter data (3C)", "Arbitrary filter data (8C)", "PERfect filtered data (0C)","PERfect filtered data (3C)","PERfect filtered data (8C)","PIME (0C)","PIME (3C)", "PIME (8C)"))tmp_samples <-c("ssu18_ps_work_0", "ssu18_ps_work_3", "ssu18_ps_work_8","ssu18_ps_filt_0", "ssu18_ps_filt_3", "ssu18_ps_filt_8","ssu18_ps_perfect_0", "ssu18_ps_perfect_3", "ssu18_ps_perfect_8","ssu18_ps_pime_0", "ssu18_ps_pime_3", "ssu18_ps_pime_8")tmp_no_samp <-c()for (i in tmp_samples) { tmp_get <-nsamples(get(i)) tmp_no_samp <-c(append(tmp_no_samp, tmp_get))}tmp_no_samp <-data.frame(tmp_no_samp)tmp_rc <-c()for (i in tmp_samples) { tmp_get <-sum(readcount(get(i))) tmp_rc <-c(append(tmp_rc, tmp_get))}tmp_rc <-data.frame(tmp_rc)tmp_asv <-c()for (i in tmp_samples) { tmp_get <-ntaxa(get(i)) tmp_asv <-c(append(tmp_asv, tmp_get))}tmp_asv <-data.frame(tmp_asv)ssu18_asv_pime_sum <- dplyr::bind_cols(tmp_objects, tmp_samples) %>% dplyr::bind_cols(., tmp_no_samp) %>% dplyr::bind_cols(., tmp_rc) %>% dplyr::bind_cols(., tmp_asv) %>% dplyr::rename("Description"=1, "object name"=2, "no. samples"=3,"total reads"=4, "total asvs"=5)rm(list =ls(pattern ="tmp_"))write.table(ssu18_asv_pime_sum, "ssu_split_by_temp.txt", sep ="\t", quote =FALSE, row.names =FALSE)``````{r}#| echo: falsessu18_alpha_ds <-c("ssu18_ps_work", "ssu18_ps_filt", "ssu18_ps_perfect", "ssu18_ps_pime")tmp_objects <-data.frame(c("FULL data set", "Arbitrary filter", "PERfect filtered","PIME filtered"))tmp_samples <-c("ssu18_ps_work","ssu18_ps_filt","ssu18_ps_perfect","ssu18_ps_pime")tmp_no_samp <-c()for (i in tmp_samples) { tmp_get <-nsamples(get(i)) tmp_no_samp <-c(append(tmp_no_samp, tmp_get))}tmp_no_samp <-data.frame(tmp_no_samp)tmp_rc <-c()for (i in tmp_samples) { tmp_get <-sum(readcount(get(i))) tmp_rc <-c(append(tmp_rc, tmp_get))}tmp_rc <-data.frame(tmp_rc)tmp_asv <-c()for (i in tmp_samples) { tmp_get <-ntaxa(get(i)) tmp_asv <-c(append(tmp_asv, tmp_get))}tmp_asv <-data.frame(tmp_asv)ssu18_asv_pime_sum <- dplyr::bind_cols(tmp_objects, tmp_samples) %>% dplyr::bind_cols(., tmp_no_samp) %>% dplyr::bind_cols(., tmp_rc) %>% dplyr::bind_cols(., tmp_asv) %>% dplyr::rename("Description"=1, "object name"=2, "no. samples"=3,"total reads"=4, "total asvs"=5)rm(list =ls(pattern ="tmp_"))write.table(ssu18_asv_pime_sum, "ssu_filter_summmary.txt", sep ="\t", quote =FALSE, row.names =FALSE)``````{r}#| echo: falsemergedGP <-merge_samples(ssu18_ps_perfect_3, "TEMP")SD <-merge_samples(sample_data(ssu18_ps_perfect_3), "TEMP")mergedGP``````{r}#| echo: falseobjects()ssu18_ps_work@phy_tree <-NULLssu18_ps_filt@phy_tree <-NULLssu18_ps_pime@phy_tree <-NULLssu18_ps_perfect@phy_tree <-NULL```We first transform all the data to relative abundance values, and compute new trees.```{r}#| code-fold: truessu18_alpha_ds <-c("ssu18_ps_work", "ssu18_ps_filt", "ssu18_ps_perfect", "ssu18_ps_pime")for (i in ssu18_alpha_ds) { tmp_ps <-transform_sample_counts(get(i), function(otu) otu/sum(otu)) tmp_ps@phy_tree <-NULL tmp_tree <-rtree(ntaxa(tmp_ps), rooted =TRUE,tip.label =taxa_names(tmp_ps)) tmp_ps_norm <-merge_phyloseq(tmp_ps, sample_data, tmp_tree) tmp_asv <-data.frame(t(otu_table(tmp_ps_norm))) tmp_ps_name <- purrr::map_chr(i, ~paste0(., "_norm"))assign(tmp_ps_name, tmp_ps_norm) tmp_asv_name <- purrr::map_chr(i, ~paste0(., "_tu"))assign(tmp_asv_name, tmp_asv)rm(list =ls(pattern ="tmp_"))}```Next, we run the analysis for all three metrics on the data sets (without a tree).```{r}#| results: hide#| code-fold: trueqvalue <-c(0,1,2)for (i in qvalue) {for (j in ssu18_alpha_ds) { tmp_asv <-get(purrr::map_chr(j, ~paste0(., "_tu"))) tmp_df <-data.frame(hill_div(tmp_asv, qvalue = i)) tmp_df <- tmp_df %>% dplyr::rename("tmp_name"=1) %>% tibble::rownames_to_column("SamName") tmp_name <- purrr::map_chr(j, ~paste0(., "_h", i))print(tmp_name)assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_")) }}objects(pattern ="_h")```And make summary tables to add back into each `ps` object.```{r}#| code-fold: truefor (i in ssu18_alpha_ds) { tmp_obs <-get(purrr::map_chr(i, ~paste0(., "_h0"))) tmp_sha <-get(purrr::map_chr(i, ~paste0(., "_h1"))) tmp_sim <-get(purrr::map_chr(i, ~paste0(., "_h2"))) tmp_hill <- dplyr::left_join(tmp_obs, tmp_sha, by ="SamName") %>% dplyr::left_join(., tmp_sim, by ="SamName") %>% dplyr::rename("Observed"=2, "Shannon_exp"=3, "InvSimpson"=4) tmp_name <- purrr::map_chr(i, ~paste0(., "_hill"))assign(tmp_name, tmp_hill)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_hill")objects()```And then create the new objects with the diversity data.```{r}#| code-fold: truefor (i in ssu18_alpha_ds) { tmp_ps <-get(i) tmp_tree <-rtree(ntaxa(tmp_ps), rooted =TRUE,tip.label =taxa_names(tmp_ps)) tmp_ps <-merge_phyloseq(tmp_ps, sample_data, tmp_tree) tmp_hill <-get(purrr::map_chr(i, ~paste0(., "_hill"))) tmp_hill_samp <- dplyr::left_join(data.frame(sample_data(tmp_ps)), tmp_hill, by ="SamName") tmp_hill_samp$ID <- tmp_hill_samp$SamName tmp_hill_samp <- tmp_hill_samp %>% tibble::column_to_rownames("ID") tmp_ps2 <-merge_phyloseq(otu_table(tmp_ps),sample_data(tmp_hill_samp),tax_table(tmp_ps),phy_tree(tmp_ps))assign(i, tmp_ps2) tmp_path <-file.path("files/alpha/rdata/")saveRDS(tmp_ps2, paste(tmp_path, i, ".rds", sep =""))rm(list =ls(pattern ="tmp_"))}objects()``````{r}#| echo: false## THIS is legacy code for using trees. Not included in this analysis#### With a phylogenetic tree## We can also run the tests using the phylogenetic tree to assess **lineage** diversity rather than ASV diversity for the filtered data sets. We do a quick check to ensure the ASV names in the ASV table and the tip names in the new phylogenetic tree are identical.ssu18_ps_pime_tree_ult <-readRDS("files/alpha/rdata/ssu18_ps_pime_tree_ult.rds")ssu18_ps_pime_otu_tree_ult <-readRDS("files/alpha/rdata/ssu18_ps_pime_otu_tree_ult.rds")ssu18_ps_perfect_tree_ult <-readRDS("files/alpha/rdata/ssu18_ps_perfect_tree_ult.rds")ssu18_ps_perfect_otu_tree_ult <-readRDS("files/alpha/rdata/ssu18_ps_perfect_otu_tree_ult.rds")ssu18_ps_filt_tree_ult <-readRDS("files/alpha/rdata/ssu18_ps_filt_tree_ult.rds")is.ultrametric(ssu18_ps_pime_tree_ult) is.ultrametric(ssu18_ps_pime_otu_tree_ult)is.ultrametric(ssu18_ps_perfect_tree_ult) is.ultrametric(ssu18_ps_perfect_otu_tree_ult)is.ultrametric(ssu18_ps_filt_tree_ult) ## To generate trees, we used the R-package [`phytools`](https://github.com/liamrevell/phytools), specifically the function `force.ultrametric` since `hill_div` requires ultrametric trees. rm(i)ssu18_alpha_ds_tree <-c("ssu18_ps_filt", "ssu18_ps_perfect", "ssu18_ps_pime")for (i in ssu18_alpha_ds_tree) { tmp_ps <-get(i) tmp_tree <- tmp_ps@phy_tree tmp_tree_ult <-force.ultrametric(tmp_tree, method =c("nnls"))print(is.ultrametric(tmp_tree_ult)) tmp_tree_name <- purrr::map_chr(i, ~paste0(., "_tree_ult"))assign(tmp_tree_name, tmp_tree_ult) tmp_path <-file.path("files/alpha/rdata/")saveRDS(tmp_tree_ult, paste(tmp_path, tmp_tree_name, ".rds", sep ="")) tmp_tmp_asv <-get(purrr::map_chr(i, ~paste0(., "_tu")))print(identical(sort(rownames(tmp_tmp_asv)), sort(tmp_tree_ult$tip.label)))rm(list =ls(pattern ="tmp_"))}objects()rm(i, j)qvalue <-c(0,1,2)for (i in qvalue) {for (j in ssu18_alpha_ds_tree) { tmp_asv <-get(purrr::map_chr(j, ~paste0(., "_tu"))) tmp_tree <-get(purrr::map_chr(j, ~paste0(., "_tree_ult"))) tmp_df <-data.frame(hill_div(tmp_asv, qvalue = i, tree = tmp_tree)) tmp_df <- tmp_df %>% dplyr::rename("tmp_name"=1) %>% tibble::rownames_to_column("SamName") tmp_name <- purrr::map_chr(j, ~paste0(., "_ht", i))print(tmp_name)assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_")) }}ssu18_alpha_ds_tree_filt <-c("ssu18_ps_filt")for (i in ssu18_alpha_ds_tree_filt) { tmp_obs <-get(purrr::map_chr(i, ~paste0(., "_ht0"))) tmp_sha <-get(purrr::map_chr(i, ~paste0(., "_ht1"))) tmp_sim <-get(purrr::map_chr(i, ~paste0(., "_ht2"))) tmp_hill <- dplyr::left_join(tmp_obs, tmp_sha, by ="SamName") %>% dplyr::left_join(., tmp_sim, by ="SamName") %>% dplyr::rename("Observed_fit"=2, "Shannon_exp_fit"=3, "InvSimpson_fit"=4) tmp_name <- purrr::map_chr(i, ~paste0(., "_hill_t"))assign(tmp_name, tmp_hill)rm(list =ls(pattern ="tmp_"))}ssu18_alpha_ds_tree_perfect <-c("ssu18_ps_perfect")for (i in ssu18_alpha_ds_tree_perfect) { tmp_obs <-get(purrr::map_chr(i, ~paste0(., "_ht0"))) tmp_sha <-get(purrr::map_chr(i, ~paste0(., "_ht1"))) tmp_sim <-get(purrr::map_chr(i, ~paste0(., "_ht2"))) tmp_hill <- dplyr::left_join(tmp_obs, tmp_sha, by ="SamName") %>% dplyr::left_join(., tmp_sim, by ="SamName") %>% dplyr::rename("Observed_pet"=2, "Shannon_exp_pet"=3, "InvSimpson_pet"=4) tmp_name <- purrr::map_chr(i, ~paste0(., "_hill_t"))assign(tmp_name, tmp_hill)rm(list =ls(pattern ="tmp_"))}ssu18_alpha_ds_tree_pime <-c("ssu18_ps_pime")for (i in ssu18_alpha_ds_tree_pime) { tmp_obs <-get(purrr::map_chr(i, ~paste0(., "_ht0"))) tmp_sha <-get(purrr::map_chr(i, ~paste0(., "_ht1"))) tmp_sim <-get(purrr::map_chr(i, ~paste0(., "_ht2"))) tmp_hill <- dplyr::left_join(tmp_obs, tmp_sha, by ="SamName") %>% dplyr::left_join(., tmp_sim, by ="SamName") %>% dplyr::rename("Observed_pit"=2, "Shannon_exp_pit"=3, "InvSimpson_pit"=4) tmp_name <- purrr::map_chr(i, ~paste0(., "_hill_t"))assign(tmp_name, tmp_hill)rm(list =ls(pattern ="tmp_"))}rm(i)for (i in ssu18_alpha_ds_tree) { tmp_ps <-get(i) tmp_hill <-get(purrr::map_chr(i, ~paste0(., "_hill_t"))) tmp_hill_samp <- dplyr::left_join(data.frame(sample_data(tmp_ps)), tmp_hill, by ="SamName") tmp_hill_samp$ID <- tmp_hill_samp$SamName tmp_hill_samp <- tmp_hill_samp %>% tibble::column_to_rownames("ID") tmp_ps2 <-merge_phyloseq(otu_table(tmp_ps),sample_data(tmp_hill_samp),tax_table(tmp_ps),phy_tree(tmp_ps))assign(i, tmp_ps2) tmp_path <-file.path("files/alpha/rdata/")saveRDS(tmp_ps2, paste(tmp_path, i, ".rds", sep =""))rm(list =ls(pattern ="tmp_"))}```## Hill Numbers SummaryNow we summarize the data for each sample against all three metrics. The table contains the results of ASV diversity estimates from the full data set and the three filtered data sets.The suffix `_fi` indicates metrics for the Arbitrary data set. The suffix `_pe` indicates metrics for the PERfect data set and the suffix `_pet` indicates the lineage diversity for the PERfect data set.The suffix `_pi` indicates metrics for the PIME data set and the suffix `_pit` indicates the lineage diversity for the PIME data set.```{r}#| echo: false######################### FULL ASVssu18_work_hill_samp <- ssu18_ps_work_hill %>% dplyr::rename("Observed"=2,"Shannon_exp"=3,"InvSimpson"=4)ssu18_work_hill_samp <- dplyr::left_join(data.frame(sample_data(ssu18_ps_work)), ssu18_work_hill_samp)ssu18_work_hill_samp <- ssu18_work_hill_samp[order(ssu18_work_hill_samp$PLOT, ssu18_work_hill_samp$TEMP),]######################### Filt ASVssu18_filt_hill_samp <- ssu18_ps_filt_hill %>% dplyr::rename("Observed_fi"=2,"Shannon_exp_fi"=3,"InvSimpson_fi"=4)######################### PERfect ASVssu18_perfect_hill_samp <- ssu18_ps_perfect_hill %>% dplyr::rename("Observed_pe"=2,"Shannon_exp_pe"=3,"InvSimpson_pe"=4)######################### PIME ASVssu18_pime_hill_samp <- ssu18_ps_pime_hill %>% dplyr::rename("Observed_pi"=2,"Shannon_exp_pi"=3,"InvSimpson_pi"=4)``````{r}#| echo: false######################### ASV TABLEssu_tab_alpha_div <- dplyr::left_join(ssu18_work_hill_samp, ssu18_filt_hill_samp) %>% dplyr::left_join(., ssu18_perfect_hill_samp) %>% dplyr::left_join(., ssu18_pime_hill_samp) %>% dplyr::relocate(c("Observed", "Observed_fi", "Observed_pe", "Observed_pi"), .after ="PAIR") %>% dplyr::relocate(c("Shannon_exp", "Shannon_exp_fi", "Shannon_exp_pe", "Shannon_exp_pi"), .after ="Observed_pi") %>% dplyr::rename("Sample_ID"="SamName") ssu_tab_alpha_div <- ssu_tab_alpha_div%>% dplyr::mutate(across(.cols =c(11:ncol(ssu_tab_alpha_div)), round, digits =1))```### Hill Summary<small>`r caption_tab_ssu("ssu_alpha_div")`</small>```{r}#| echo: false#| eval: trueseq_table <- ssu_tab_alpha_divseq_table[2:6] <-NULLwrite.table(seq_table, "files/alpha/tables/ssu_tab_alpha_div.txt", row.names =FALSE, sep ="\t", quote =FALSE)``````{r}#| echo: false#| eval: truereactable(seq_table,columns =list(Sample_ID =colDef(name ="Sample_ID", sticky ="left", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee"), align ="left",minWidth =150),Observed =colDef(name ="FULL"),Observed_fi =colDef(name ="Arbitrary"),Observed_pe =colDef(name ="PERfect"),Observed_pi =colDef(name ="PIME", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee")),Shannon_exp =colDef(name ="FULL"),Shannon_exp_fi =colDef(name ="Arbitrary"),Shannon_exp_pe =colDef(name ="PERfect"),Shannon_exp_pi =colDef(name ="PIME", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee")),InvSimpson =colDef(name ="FULL"),InvSimpson_fi =colDef(name ="Arbitrary"),InvSimpson_pe =colDef(name ="PERfect"),InvSimpson_pi =colDef(name ="PIME") ),columnGroups =list(colGroup(name ="Observed richness", columns =c("Observed", "Observed_fi", "Observed_pe", "Observed_pi"),headerStyle =list(borderRight ="5px solid #eee", fontSize ="1.1em")),colGroup(name ="Shannon exponential", columns =c("Shannon_exp", "Shannon_exp_fi", "Shannon_exp_pe", "Shannon_exp_pi"),headerStyle =list(fontSize ="1.1em")),colGroup(name ="Simpson multiplicative inverse", columns =c("InvSimpson", "InvSimpson_fi", "InvSimpson_pe", "InvSimpson_pi"),headerStyle =list(fontSize ="1.1em")) ),defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =0),align ="center", filterable =TRUE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold"), minWidth =100 ), searchable =TRUE, defaultPageSize =5, showPagination =TRUE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =FALSE, showSortable =TRUE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```{{< downloadthis files/alpha/tables/ssu_tab_alpha_div.txt dname="ssu_tab_alpha_div" label="Download table as text file" icon="table" type="primary" >}}## Normality TestsBefore running significance tests, we need to know if data is normally distributed, which will tell use whether to use a parametric or non-parametric test. To test if the data are normally distributed, we use the Shapiro-Wilk Normality test and the Bartlett Test of Homogeneity of Variances.If the p-values are both **not significant** (p > 0.05) from the tests, we accept the null hypothesis (that the results are normally distributed) and test for significance between samples using an ANOVA. If the p-values are both **significant** (p < 0.05), we reject the null hypothesis (that the results are normally distributed) and test for significance between samples using Kruskal-Wallis (non-parametric equivalent of ANOVA).The commands are as follows:`shapiro.test(x)`, where `x` is a numeric vector of alpha diversity values from the sample data table.`bartlett.test(Value ~ Group, data = df)` Where `Value` is the metric of interest, `Group` in the treatment to compare, and `df` is the data frame. First the Shapiro-Wilk Normality test.```{r}#| code-fold: truessu_tab_alpha_divssu18_div_tab <- ssu_tab_alpha_divssu18_shap_tests_asv <-c()for (i incolnames(ssu18_div_tab[,7:ncol(ssu18_div_tab)])) { tmp_name <- purrr::map_chr(i, ~paste0("ssu18_shap_asv_", .)) ssu18_shap_tests_asv <-append(ssu18_shap_tests_asv, tmp_name) tmp_test <-eval(shapiro.test(ssu18_div_tab[[i]])) tmp_test$data.name <- tmp_nameassign(tmp_name, tmp_test)rm(list =ls(pattern ="tmp_"))}objects(pattern ="shap_")```And then the Bartlett Test of Homogeneity of Variances.```{r}#| code-fold: truessu18_div_tab_asv <- ssu_tab_alpha_divssu18_bart_tests_asv <-c()for (i incolnames(ssu18_div_tab_asv[,7:ncol(ssu18_div_tab_asv)])) { tmp_name <- purrr::map_chr(i, ~paste0("ssu18_bart_asv_", .)) ssu18_bart_tests_asv <-append(ssu18_bart_tests_asv, tmp_name) tmp_test <-eval(bartlett.test(ssu18_div_tab_asv[[i]] ~ TEMP, data = ssu18_div_tab_asv)) tmp_test$data.name <- tmp_nameassign(tmp_name, tmp_test)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bart_")```Here we see which Shapiro-Wilk Normality **and** Bartlett tests were significant and which were not. So wherever the value of both p-values in `> 0.05` we can use an ANOVA, otherwise we use Kruskal-Wallis.```{r}#| echo: false#| eval: true#| warning: false#| message: falsessu18_norm_res <-data.frame()for (i incolnames(ssu18_div_tab_asv[,7:ncol(ssu18_div_tab_asv)])) { tmp_get_shap <-get(purrr::map_chr(i, ~paste0("ssu18_shap_asv_", .))) tmp_shap_p <-round(tmp_get_shap[[2]], 4) tmp_get_bart <-get(purrr::map_chr(i, ~paste0("ssu18_bart_asv_", .))) tmp_bart_p <-round(tmp_get_bart[[3]], 4) tmp_test <-if(tmp_shap_p >0.05& tmp_bart_p >0.05 ){ tmp_test <-"ANOVA" } else { tmp_test <-"Kruskal-Wallis" } tmp_df <-data.frame(i, tmp_shap_p, tmp_bart_p, tmp_test) ssu18_norm_res <- dplyr::bind_rows(ssu18_norm_res, tmp_df)rm(list =ls(pattern ="tmp_"))}ssu18_norm_res[,1] <-gsub('_exp', '', ssu18_norm_res[,1])ssu18_norm_res <- ssu18_norm_res %>%separate(col =1, into =c("metric", "dataset"), sep ="_", remove =TRUE)ssu18_norm_res <- ssu18_norm_res %>% tidyr::replace_na(list(dataset ="FULL"))ssu18_norm_res$dataset <- stringr::str_replace(ssu18_norm_res$dataset, "^fi$", "FILT") %>% stringr::str_replace(., "^pi$", "PIME") %>% stringr::str_replace(., "^pe$", "PERfect")ssu18_norm_res <- ssu18_norm_res %>% dplyr::rename(c("p-value (normality)"=3, "p-value (homogeneity)"=4, "method"=5))```<small>`r caption_tab_ssu("ssu_norm_tests")`</small>```{r}#| echo: false#| eval: trueseq_table <- ssu18_norm_reswrite.table(ssu18_norm_res, "files/alpha/tables/ssu_norm_tests.txt", row.names =FALSE, sep ="\t", quote =FALSE)``````{r}#| echo: false#| eval: truereactable(seq_table,defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =0),align ="center", filterable =FALSE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold"), minWidth =100, headerVAlign ="center" ), columns =list(metric =colDef(name ="Metric", sticky ="left", style =list(borderRight ="1px solid #eee"),headerStyle =list(borderRight ="1px solid #eee"), align ="left",minWidth =100),`p-value (normality)`=colDef(name ="p-value (normality)", minWidth =100, resizable =FALSE,style =function(value) {if (value >0.05) { color <-"#2271B2" } elseif (value <0.05) { color <-"#B22271" } else { color <-"#777" }list(color = color, fontWeight ="bold") }),`p-value (homogeneity)`=colDef(name ="p-value (homogeneity)", minWidth =130, resizable =FALSE,style =function(value) {if (value >0.05) { color <-"#2271B2"#green "#", "#" } elseif (value <0.05) { color <-"#B22271"#RED } else { color <-"#777" }list(color = color, fontWeight ="bold") }),`dataset`=colDef(name ="dataset", minWidth =80, resizable =FALSE),`method`=colDef(name ="method", minWidth =120, resizable =FALSE) ), searchable =FALSE, defaultPageSize =12, showPagination =FALSE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =TRUE, showSortable =FALSE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```{{< downloadthis files/alpha/tables/ssu_norm_tests.txt dname="ssu_norm_tests" label="Download table as text file" icon="table" type="primary" >}}## Significance TestsTo begin, we need to create a hierarchy variable; a two-column matrix specifying the relationship between samples (first column) and groups (second column).```{r}#| code-fold: truessu18_hill_hier <- ssu_tab_alpha_divssu18_hill_hier <- ssu18_hill_hier %>% dplyr::select("Sample_ID", "TEMP") %>% tibble::remove_rownames()ssu18_hill_hier <- ssu18_hill_hier[order(ssu18_hill_hier$TEMP), ]ssu18_hill_hier$TEMP =paste(ssu18_hill_hier$TEMP, 'C', sep='')ssu18_hill_hier <- ssu18_hill_hier %>% tibble::remove_rownames()saveRDS(ssu18_hill_hier, "files/alpha/rdata/ssu18_hill_hier.rds")```Again, we start by testing significance of ASV diversity for the data sets against each of the three metrics using the `div_test` function.The command is as follows:```div_test(countable = x, qvalue = i, hierarchy = hier, tree = ultrametric_tree, posthoc = TRUE)```where `x` is ASV by sample table, `i` is the q-value corresponding to the metric of interest, `hier` is the hierarchy matrix, `tree` is an ultrametric formatted phylogenetic tree (if you choose to explore lineage diversity), and `posthoc` indicates whether to run post hoc pairwise analyses.```{r}#| results: hide#| code-fold: trueqvalue <-c(0,1,2)for (i in ssu18_alpha_ds) {for (j in qvalue) { tmp_get <-get(purrr::map_chr(i, ~paste0(., "_tu"))) tmp_test <-div_test(tmp_get, qvalue = j,hierarchy = ssu18_hill_hier,posthoc =TRUE) tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))print(tmp_name)assign(tmp_name, tmp_test)rm(list =ls(pattern ="tmp_")) }}``````{r}#| echo: false## Legacy code for lineage diversity## And then test significance of lineage diversity for the filtered data sets.ssu18_alpha_ds_tree <-c("ssu18_ps_filt", "ssu18_ps_perfect", "ssu18_ps_pime")qvalue <-c(0,1,2)for (i in ssu18_alpha_ds_tree) {for (j in qvalue) { tmp_get <-get(purrr::map_chr(i, ~paste0(., "_tu"))) tmp_tree <-get(purrr::map_chr(i, ~paste0(., "_tree_ult"))) tmp_test <-div_test(tmp_get, qvalue = j,hierarchy = ssu18_hill_hier,posthoc =TRUE,tree = tmp_tree) tmp_name <- purrr::map_chr(i, ~paste0(., "_t_q", j, "_adt"))print(tmp_name)assign(tmp_name, tmp_test)rm(list =ls(pattern ="tmp_")) }}``````{r}#| echo: falsetmp_objects <-c("ssu18_ps_work", "ssu18_ps_filt", "ssu18_ps_perfect", "ssu18_ps_pime")tmp_metric <-data.frame(c("Observed", "Shannon exponential", "Inverse Simpson"))tmp_qvalue <-data.frame(c("0", "1", "2"))qvalue <-c(0,1,2)for (i in tmp_objects) { tmp_h_pvalue <-c()for (j in qvalue) { tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt")) tmp_get <-get(tmp_name)$homogeneity.pvalue tmp_h_pvalue <-c(append(tmp_h_pvalue, tmp_get)) } tmp_h_pvalue <-data.frame(tmp_h_pvalue) tmp_n_pvalue <-c()for (j in qvalue) { tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt")) tmp_get <-get(tmp_name)$normality.pvalue tmp_n_pvalue <-c(append(tmp_n_pvalue, tmp_get)) } tmp_n_pvalue <-data.frame(tmp_n_pvalue) tmp_method <-c()for (j in qvalue) { tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt")) tmp_get <-get(tmp_name)$method tmp_method <-c(append(tmp_method, tmp_get)) } tmp_method <-data.frame(tmp_method) tmp_phoc_method <-c()for (j in qvalue) { tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt")) tmp_get <-get(tmp_name)$posthoc.method tmp_phoc_method <-c(append(tmp_phoc_method, tmp_get)) } tmp_phoc_method <-data.frame(tmp_phoc_method) tmp_df <- dplyr::bind_cols(tmp_metric, tmp_qvalue) %>% dplyr::bind_cols(., tmp_n_pvalue) %>% dplyr::bind_cols(., tmp_h_pvalue) %>% dplyr::bind_cols(., tmp_method) %>% dplyr::bind_cols(., tmp_phoc_method) %>% dplyr::rename("metric"=1, "q-value"=2, "normality p-value"=3, "homogeneity p-value"=4, "method"=5, "posthoc method"=6) tmp_name <- purrr::map_chr(i, ~paste0(i, "_sig_tab"))assign(tmp_name, tmp_df)}``````{r}#| echo: false## Format as follows:## FALSE Kruskal-Wallis Test = its18_pime_q1_adt$test[[3]]## TRUE Tukey post-hoc = its18_ps_work_q0_adt$test[[1]][[1, 5]]############################### WORK ##################################################tmp_pvalue <-data.frame(c(ssu18_ps_work_q0_adt$test[[1]][[1, 5]], ssu18_ps_work_q1_adt$test[[1]][[1, 5]], ssu18_ps_work_q2_adt$test[[1]][[1, 5]]))ssu18_ps_work_sig_tab <- dplyr::bind_cols(ssu18_ps_work_sig_tab, tmp_pvalue) %>% dplyr::rename("posthoc p-value"=7)####################################Filt#############################################tmp_pvalue <-data.frame(c(ssu18_ps_filt_q0_adt$test[[1]][[1, 5]], ssu18_ps_filt_q1_adt$test[[1]][[1, 5]], ssu18_ps_filt_q2_adt$test[[1]][[1, 5]]))ssu18_ps_filt_sig_tab <- dplyr::bind_cols(ssu18_ps_filt_sig_tab, tmp_pvalue) %>% dplyr::rename("posthoc p-value"=7)####################################PERfect##########################################tmp_pvalue <-data.frame(c(ssu18_ps_perfect_q0_adt$test[[1]][[1, 5]], ssu18_ps_perfect_q1_adt$test[[1]][[1, 5]], ssu18_ps_perfect_q2_adt$test[[1]][[1, 5]]))ssu18_ps_perfect_sig_tab <- dplyr::bind_cols(ssu18_ps_perfect_sig_tab, tmp_pvalue) %>% dplyr::rename("posthoc p-value"=7)################################ PIME ################################################tmp_pvalue <-data.frame(c(ssu18_ps_pime_q0_adt$test[[1]][[1, 5]], ssu18_ps_pime_q1_adt$test[[3]], ssu18_ps_pime_q2_adt$test[[1]][[1, 5]]))ssu18_ps_pime_sig_tab <- dplyr::bind_cols(ssu18_ps_pime_sig_tab, tmp_pvalue) %>% dplyr::rename("posthoc p-value"=7)rm(list =ls(pattern ="tmp_"))``````{r}#| echo: falsessu18_ps_work_sig_tab$dataset <-"FULL"ssu18_ps_work_sig_tab$type <-"ASV"ssu18_ps_work_sig_tab$lineage <-"No"ssu18_ps_work_sig_tab <- ssu18_ps_work_sig_tab[,c(8:10,1:7)]ssu18_ps_pime_sig_tab$dataset <-"PIME"ssu18_ps_pime_sig_tab$type <-"ASV"ssu18_ps_pime_sig_tab$lineage <-"No"ssu18_ps_pime_sig_tab <- ssu18_ps_pime_sig_tab[,c(8:10,1:7)]ssu18_ps_perfect_sig_tab$dataset <-"PERfect"ssu18_ps_perfect_sig_tab$type <-"ASV"ssu18_ps_perfect_sig_tab$lineage <-"No"ssu18_ps_perfect_sig_tab <- ssu18_ps_perfect_sig_tab[,c(8:10,1:7)]ssu18_ps_filt_sig_tab$dataset <-"FILT"ssu18_ps_filt_sig_tab$type <-"ASV"ssu18_ps_filt_sig_tab$lineage <-"No"ssu18_ps_filt_sig_tab <- ssu18_ps_filt_sig_tab[,c(8:10,1:7)]ssu18_sig_tab_all <-rbind(ssu18_ps_work_sig_tab, ssu18_ps_filt_sig_tab, ssu18_ps_pime_sig_tab, ssu18_ps_perfect_sig_tab )```## Summary<small>`r caption_tab_ssu("ssu_sign_tests")`</small>```{r}#| echo: false#| eval: true#| warning: false#| message: falseseq_table <- ssu18_sig_tab_allseq_table[,2:3] <-NULLseq_table <- seq_table %>% dplyr::mutate(across(.cols =c(4:5), round, digits =4)) write.table(seq_table, "files/alpha/tables/ssu_sign_tests.txt", row.names =FALSE, sep ="\t", quote =FALSE)``````{r}#| echo: false#| eval: truereactable(seq_table,defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =0),align ="center", filterable =FALSE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold"), minWidth =100 ), columns =list(dataset =colDef(name ="Dataset", sticky ="left", style =list(borderRight ="1px solid #eee"),headerStyle =list(borderRight ="1px solid #eee"), align ="left",minWidth =80),`posthoc p-value`=colDef(name ="posthoc p-value", style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color, fontWeight ="bold") }, cell =function(value) format(value, digits =3, scientific =TRUE) ),metric =colDef(name ="metric", minWidth =140, align ="left"),`q-value`=colDef(name ="q-value", minWidth =70, align ="center"),`normality p-value`=colDef(name ="p-value (normality)", minWidth =100,),`homogeneity p-value`=colDef(name ="p-value (homogeneity)", minWidth =130),`dataset`=colDef(name ="dataset", minWidth =80),`method`=colDef(name ="method", minWidth =120) ), searchable =FALSE, defaultPageSize =12, showPagination =FALSE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =FALSE, showSortable =FALSE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.7em")))#rm(seq_table)```{{< downloadthis files/alpha/tables/ssu_sign_tests.txt dname="ssu_sign_tests" label="Download table as text file" icon="table" type="primary" >}}## PostHoc Analyses First let's check the results of each posthoc analysis.::: {.column-body}::: {.panel-tabset}### Observed (q-value = 0)```{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvf0_lab <-"FULL (Observed)"ssu18_asvf0_labssu18_ps_work_q0_adt$posthoc.methoddata.frame(ssu18_ps_work_q0_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvl0_lab <-"FILT (Observed)"ssu18_asvl0_labssu18_ps_filt_q0_adt$posthoc.methoddata.frame(ssu18_ps_filt_q0_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvp0_lab <-"PIME (Observed)"ssu18_asvp0_labssu18_ps_pime_q0_adt$posthoc.methoddata.frame(ssu18_ps_pime_q0_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvr0_lab <-"PERfect (Observed)"ssu18_asvr0_labssu18_ps_perfect_q0_adt$posthoc.methoddata.frame(ssu18_ps_perfect_q0_adt$posthoc)```### Shannon exp (q-value = 1)```{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvf1_lab <-"FULL (Shannon exponential)"ssu18_asvf1_labssu18_ps_work_q1_adt$posthoc.methoddata.frame(ssu18_ps_work_q1_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvl1_lab <-"FILT (Shannon exponential)"ssu18_asvl1_labssu18_ps_filt_q1_adt$posthoc.methoddata.frame(ssu18_ps_filt_q1_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvp1_lab <-"PIME (Shannon exponential)"ssu18_asvp1_labssu18_ps_pime_q1_adt$posthoc.methoddata.frame(ssu18_ps_pime_q1_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvr1_lab <-"PERfect (Shannon exponential)"ssu18_asvr1_labssu18_ps_perfect_q1_adt$posthoc.methoddata.frame(ssu18_ps_perfect_q1_adt$posthoc)```### Inverse Simpson (q-value = 2)```{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvf2_lab <-"FULL (Inverse Simpson)"ssu18_asvf2_labssu18_ps_work_q2_adt$posthoc.methoddata.frame(ssu18_ps_work_q2_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvl2_lab <-"FILT (Inverse Simpson)"ssu18_asvl2_labssu18_ps_filt_q2_adt$posthoc.methoddata.frame(ssu18_ps_filt_q2_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvp2_lab <-"PIME (Inverse Simpson)"ssu18_asvp2_labssu18_ps_pime_q2_adt$posthoc.methoddata.frame(ssu18_ps_pime_q2_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' ssu18_asvr2_lab <-"PERfect (Inverse Simpson)"ssu18_asvr2_labssu18_ps_perfect_q2_adt$posthoc.methoddata.frame(ssu18_ps_perfect_q2_adt$posthoc)```::::::Now we can plot the results from the posthoc analyses for each metric and data set using the function `div_test_plot_jjs`. I modified the original function (`div_test_plot`) to control a little of the formatting.The command is as follows:```div_test_plot(divtest = x, chart = "type", colour = col.pal, posthoc = TRUE, threshold = value))```where `x` is the results from the `div_test` function, `"type"` is chart type (box, jitter, or violin), `colour` is is a color palette, `posthoc` indicates whether to run posthoc pairwise analyses, and `value` is the maximum p-value to show in pairwise posthoc results. **WARNING** if none of the posthoc results are below the specified threshold, the function will throw an error. Therefore, until this is fixed, all posthoc values are shown.```{r}#| code-fold: truesource("hack_code/div_test_plot_jjs.R")rm(list =ls(pattern ="_adt_plot"))for (i inobjects(pattern ="_adt")) { tmp_name <- purrr::map_chr(i, ~paste0(., "_plot")) tmp_get <-get(i) tmp_df <-div_test_plot_jjs(tmp_get, chart ="box",colour = swel_col, posthoc =TRUE) tmp_df <-ggpar(tmp_df, legend ="none")print(tmp_name)assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_"))}``````{r}#| echo: false#| eval: falsessu18_ps_work_q0_adt_plot <- ssu18_ps_work_q0_adt_plot +theme(axis.title.x =element_blank()) +ggtitle(ssu18_asvf0_lab) +theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_filt_q0_adt_plot <- ssu18_ps_filt_q0_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(ssu18_asvl0_lab) +theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_perfect_q0_adt_plot <- ssu18_ps_perfect_q0_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(ssu18_asvr0_lab) +theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_pime_q0_adt_plot <- ssu18_ps_pime_q0_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(ssu18_asvp0_lab)+theme(plot.title =element_text(size =20, face ="bold"))#####################ssu18_ps_work_q1_adt_plot <- ssu18_ps_work_q1_adt_plot +theme(axis.title.x =element_blank()) +ggtitle(ssu18_asvf1_lab)+theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_filt_q1_adt_plot <- ssu18_ps_filt_q1_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(ssu18_asvl1_lab) +theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_perfect_q1_adt_plot <- ssu18_ps_perfect_q1_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(ssu18_asvr1_lab) +theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_pime_q1_adt_plot <- ssu18_ps_pime_q1_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(ssu18_asvp1_lab) +theme(plot.title =element_text(size =20, face ="bold"))#####################ssu18_ps_work_q2_adt_plot <- ssu18_ps_work_q2_adt_plot +ggtitle(ssu18_asvf2_lab) +theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_filt_q2_adt_plot <- ssu18_ps_filt_q2_adt_plot +theme(axis.title.y =element_blank()) +ggtitle(ssu18_asvl2_lab) +theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_perfect_q2_adt_plot <- ssu18_ps_perfect_q2_adt_plot +theme(axis.title.y =element_blank()) +ggtitle(ssu18_asvr2_lab) +theme(plot.title =element_text(size =20, face ="bold"))ssu18_ps_pime_q2_adt_plot <- ssu18_ps_pime_q2_adt_plot +theme(axis.title.y =element_blank()) +ggtitle(ssu18_asvp2_lab) +theme(plot.title =element_text(size =20, face ="bold"))ssu18_alph_div_plots_asv <-ggarrange( ssu18_ps_work_q0_adt_plot, ssu18_ps_filt_q0_adt_plot, ssu18_ps_perfect_q0_adt_plot, ssu18_ps_pime_q0_adt_plot, ssu18_ps_work_q1_adt_plot, ssu18_ps_filt_q1_adt_plot, ssu18_ps_perfect_q1_adt_plot, ssu18_ps_pime_q1_adt_plot, ssu18_ps_work_q2_adt_plot, ssu18_ps_filt_q2_adt_plot, ssu18_ps_perfect_q2_adt_plot, ssu18_ps_pime_q2_adt_plot, ncol =4, nrow =3)ssu18_alph_div_plots_asvdev.off()png("files/alpha/figures/ssu18_alph_div_plots_asv.png",height =32, width =60, units ='cm', res =600, bg ="white")ssu18_alph_div_plots_asvdev.off()pdf("files/alpha/figures/ssu18_alph_div_plots_asv.pdf",height =10, width =11)ssu18_alph_div_plots_asvdev.off()system("cp files/alpha/figures/ssu18_alph_div_plots_asv.png include/alpha/ssu18_alph_div_plots_asv.png")```## Alpha Diversity PlotsPosthoc adjusted p-values given for each pairwise comparison.```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 5#| column: body-outsetknitr::include_graphics("include/alpha/ssu18_alph_div_plots_asv.png")```<small>`r caption_fig_ssu("ssu_alpha_div_plots")`</small>```{r}#| echo: falsesave.image("page_build/alpha_ssu18_alpha_wf.rdata")``````{r}#| echo: falsesamp_ps <-c("ssu18_ps_work", "ssu18_ps_filt", "ssu18_ps_pime", "ssu18_ps_perfect")for (i in samp_ps) {dir.create(paste("files/alpha/figures/ssu/", i, sep =""), recursive =TRUE) tmp_get_q0 <-get(paste0(i, "_q0_adt_plot", sep ="")) tmp_get_q1 <-get(paste0(i, "_q1_adt_plot", sep ="")) tmp_get_q2 <-get(paste0(i, "_q2_adt_plot", sep ="")) ggplot2::ggsave(tmp_get_q0, file =paste0("files/alpha/figures/ssu/", i, "/", i, "_q0_adt_plot", ".png", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q0, file =paste0("files/alpha/figures/ssu/", i, "/", i, "_q0_adt_plot", ".pdf", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q1, file =paste0("files/alpha/figures/ssu/", i, "/", i, "_q1_adt_plot", ".png", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q1, file =paste0("files/alpha/figures/ssu/", i, "/", i, "_q1_adt_plot", ".pdf", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q2, file =paste0("files/alpha/figures/ssu/", i, "/", i, "_q2_adt_plot", ".png", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q2, file =paste0("files/alpha/figures/ssu/", i, "/", i, "_q2_adt_plot", ".pdf", sep =""), width =5, height =3)rm(list =ls(pattern ="tmp_"))} ```# ITS```{r}#| include: false## Initial Load for ANALYSIS #1set.seed(119)## ASV FULL AND PIME AND PERfectits18_ps_work <-readRDS("files/data-prep/rdata/its18_ps_work.rds")its18_ps_filt <-readRDS("files/filtering/arbitrary/rdata/its18_ps_filt.rds")its18_ps_perfect <-readRDS("files/filtering/perfect/rdata/its18_ps_perfect.rds")its18_ps_pime <-readRDS("files/filtering/pime/rdata/its18_ps_pime.rds")``````{r}#| echo: falseswel_col <-c("#2271B2", "#71B222", "#B22271")```To account for presence of rare sequence variants caused by sequencing errors or other technical artifacts, we use Hill numbers [@alberdi2019guide]. Hill numbers allow the weight put on rare versus abundant sequence variants to be scaled while providing intuitive comparisons of diversity levels using “effective number of ASVs” as a measuring unit. This approach allows for balancing the over representation of rare ASVs that might be inflated due to sequencing errors.We will then use Shapiro-Wilk tests to test for normalcy and then, depending on the results, either use parametric ANOVA or non-parametric Kruskal-Wallis to compare alpha diversity among treatments.## Calculate Hill NumbersTo calculate Hill numbers, we use the R package `hilldiv`[@alberdi2019hilldiv]. We calculate three metrics that put more or less weight on common species:1) Observed richness, where `q-value = 0`.2) Shannon exponential, which weighs ASVs by their frequency, where `q-value = 1`.3) Simpson multiplicative inverse, which over weighs abundant ASVs, where `q-value = 2`.We perform each analysis against the Full (unfiltered) data set as well as the Arbitrary, PERfect and PIME, filtered data sets using the function `hill_div`.The command is as follows:```hill_div(count = x, qvalue = i, tree = ultrametric_tree)```where `x` is the sample by ASV table, `i` is the q-value corresponding to the metric of interest and `tree` is an ultrametric formatted phylogenetic tree, however this really doesn't apply for ITS data anyway.We first transform all the data to relative abundance values, and compute new trees.```{r}#| code-fold: trueits18_alpha_ds <-c("its18_ps_work", "its18_ps_filt", "its18_ps_perfect", "its18_ps_pime")for (i in its18_alpha_ds) { tmp_ps <-transform_sample_counts(get(i), function(otu) otu/sum(otu)) tmp_ps@phy_tree <-NULL tmp_tree <-rtree(ntaxa(tmp_ps), rooted =TRUE,tip.label =taxa_names(tmp_ps)) tmp_ps_norm <-merge_phyloseq(tmp_ps, sample_data, tmp_tree) tmp_asv <-data.frame(t(otu_table(tmp_ps_norm))) tmp_ps_name <- purrr::map_chr(i, ~paste0(., "_norm"))assign(tmp_ps_name, tmp_ps_norm) tmp_asv_name <- purrr::map_chr(i, ~paste0(., "_tu"))assign(tmp_asv_name, tmp_asv)rm(list =ls(pattern ="tmp_"))}```Next, we run the analysis for all three metrics on the data sets (without a tree).```{r}#| results: hide#| code-fold: trueqvalue <-c(0,1,2)for (i in qvalue) {for (j in its18_alpha_ds) { tmp_asv <-get(purrr::map_chr(j, ~paste0(., "_tu"))) tmp_df <-data.frame(hill_div(tmp_asv, qvalue = i)) tmp_df <- tmp_df %>% dplyr::rename("tmp_name"=1) %>% tibble::rownames_to_column("SamName") tmp_name <- purrr::map_chr(j, ~paste0(., "_h", i))print(tmp_name)assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_")) }}objects(pattern ="_h")```And make summary tables to add back into each `ps` object.```{r}#| code-fold: truefor (i in its18_alpha_ds) { tmp_obs <-get(purrr::map_chr(i, ~paste0(., "_h0"))) tmp_sha <-get(purrr::map_chr(i, ~paste0(., "_h1"))) tmp_sim <-get(purrr::map_chr(i, ~paste0(., "_h2"))) tmp_hill <- dplyr::left_join(tmp_obs, tmp_sha, by ="SamName") %>% dplyr::left_join(., tmp_sim, by ="SamName") %>% dplyr::rename("Observed"=2, "Shannon_exp"=3, "InvSimpson"=4) tmp_name <- purrr::map_chr(i, ~paste0(., "_hill"))assign(tmp_name, tmp_hill)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_hill")objects()```And then create the new objects with the diversity data.```{r}#| code-fold: truefor (i in its18_alpha_ds) { tmp_ps <-get(i) tmp_tree <-rtree(ntaxa(tmp_ps), rooted =TRUE,tip.label =taxa_names(tmp_ps)) tmp_ps <-merge_phyloseq(tmp_ps, sample_data, tmp_tree) tmp_hill <-get(purrr::map_chr(i, ~paste0(., "_hill"))) tmp_hill_samp <- dplyr::left_join(data.frame(sample_data(tmp_ps)), tmp_hill, by ="SamName") tmp_hill_samp$ID <- tmp_hill_samp$SamName tmp_hill_samp <- tmp_hill_samp %>% tibble::column_to_rownames("ID") tmp_ps2 <-merge_phyloseq(otu_table(tmp_ps),sample_data(tmp_hill_samp),tax_table(tmp_ps),phy_tree(tmp_ps))assign(i, tmp_ps2) tmp_path <-file.path("files/alpha/rdata/")saveRDS(tmp_ps2, paste(tmp_path, i, ".rds", sep =""))rm(list =ls(pattern ="tmp_"))}objects()```## Hill Numbers SummaryNow we summarize the data for each sample against all three metrics. The table contains the results of ASV diversity estimates from the full data set and the three filtered data sets.The suffix `_fi` indicates metrics for the Arbitrary data set. The suffix `_pe` indicates metrics for the PERfect data set and the suffix `_pet` indicates the lineage diversity for the PERfect data set.The suffix `_pi` indicates metrics for the PIME data set and the suffix `_pit` indicates the lineage diversity for the PIME data set.```{r}#| echo: false######################### FULL ASVits18_work_hill_samp <- its18_ps_work_hill %>% dplyr::rename("Observed"=2,"Shannon_exp"=3,"InvSimpson"=4)its18_work_hill_samp <- dplyr::left_join(data.frame(sample_data(its18_ps_work)), its18_work_hill_samp)its18_work_hill_samp <- its18_work_hill_samp[order(its18_work_hill_samp$PLOT, its18_work_hill_samp$TEMP),]######################### Filt ASVits18_filt_hill_samp <- its18_ps_filt_hill %>% dplyr::rename("Observed_fi"=2,"Shannon_exp_fi"=3,"InvSimpson_fi"=4)######################### PERfect ASVits18_perfect_hill_samp <- its18_ps_perfect_hill %>% dplyr::rename("Observed_pe"=2,"Shannon_exp_pe"=3,"InvSimpson_pe"=4)######################### PIME ASVits18_pime_hill_samp <- its18_ps_pime_hill %>% dplyr::rename("Observed_pi"=2,"Shannon_exp_pi"=3,"InvSimpson_pi"=4)``````{r}#| echo: false######################### ASV TABLEits_tab_alpha_div <- dplyr::left_join(its18_work_hill_samp, its18_filt_hill_samp) %>% dplyr::left_join(., its18_perfect_hill_samp) %>% dplyr::left_join(., its18_pime_hill_samp) %>% dplyr::relocate(c("Observed", "Observed_fi", "Observed_pe", "Observed_pi"), .after ="PAIR") %>% dplyr::relocate(c("Shannon_exp", "Shannon_exp_fi", "Shannon_exp_pe", "Shannon_exp_pi"), .after ="Observed_pi") %>% dplyr::rename("Sample_ID"="SamName") its_tab_alpha_div <- its_tab_alpha_div%>% dplyr::mutate(across(.cols =c(11:ncol(its_tab_alpha_div)), round, digits =1))```### Hill Summary<small>`r caption_tab_its("its_alpha_div")`</small>```{r}#| echo: false#| eval: trueseq_table <- its_tab_alpha_divseq_table[2:6] <-NULLwrite.table(seq_table, "files/alpha/tables/its_tab_alpha_div.txt", row.names =FALSE, sep ="\t", quote =FALSE)``````{r}#| echo: false#| eval: truereactable(seq_table,columns =list(Sample_ID =colDef(name ="Sample_ID", sticky ="left", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee"), align ="left",minWidth =150),Observed =colDef(name ="FULL"),Observed_fi =colDef(name ="Arbitrary"),Observed_pe =colDef(name ="PERfect"),Observed_pi =colDef(name ="PIME", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee")),Shannon_exp =colDef(name ="FULL"),Shannon_exp_fi =colDef(name ="Arbitrary"),Shannon_exp_pe =colDef(name ="PERfect"),Shannon_exp_pi =colDef(name ="PIME", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee")),InvSimpson =colDef(name ="FULL"),InvSimpson_fi =colDef(name ="Arbitrary"),InvSimpson_pe =colDef(name ="PERfect"),InvSimpson_pi =colDef(name ="PIME") ),columnGroups =list(colGroup(name ="Observed richness", columns =c("Observed", "Observed_fi", "Observed_pe", "Observed_pi"),headerStyle =list(borderRight ="5px solid #eee", fontSize ="1.1em")),colGroup(name ="Shannon exponential", columns =c("Shannon_exp", "Shannon_exp_fi", "Shannon_exp_pe", "Shannon_exp_pi"),headerStyle =list(fontSize ="1.1em")),colGroup(name ="Simpson multiplicative inverse", columns =c("InvSimpson", "InvSimpson_fi", "InvSimpson_pe", "InvSimpson_pi"),headerStyle =list(fontSize ="1.1em")) ),defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =0),align ="center", filterable =TRUE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold"), minWidth =100 ), searchable =TRUE, defaultPageSize =5, showPagination =TRUE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =FALSE, showSortable =TRUE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```{{< downloadthis files/alpha/tables/its_tab_alpha_div.txt dname="its_tab_alpha_div" label="Download table as text file" icon="table" type="primary" >}}## Normality TestsBefore running significance tests, we need to know if data is normally distributed, which will tell use whether to use a parametric or non-parametric test. To test if the data are normally distributed, we use the Shapiro-Wilk Normality test and the Bartlett Test of Homogeneity of Variances.If the p-values are both **not significant** (p > 0.05) from the tests, we accept the null hypothesis (that the results are normally distributed) and test for significance between samples using an ANOVA. If the p-values are both **significant** (p < 0.05), we reject the null hypothesis (that the results are normally distributed) and test for significance between samples using Kruskal-Wallis (non-parametric equivalent of ANOVA).The commands are as follows:`shapiro.test(x)`, where `x` is a numeric vector of alpha diversity values from the sample data table.`bartlett.test(Value ~ Group, data = df)` Where `Value` is the metric of interest, `Group` in the treatment to compare, and `df` is the data frame. First the Shapiro-Wilk Normality test.```{r}#| code-fold: trueits_tab_alpha_divits18_div_tab <- its_tab_alpha_divits18_shap_tests_asv <-c()for (i incolnames(its18_div_tab[,7:ncol(its18_div_tab)])) { tmp_name <- purrr::map_chr(i, ~paste0("its18_shap_asv_", .)) its18_shap_tests_asv <-append(its18_shap_tests_asv, tmp_name) tmp_test <-eval(shapiro.test(its18_div_tab[[i]])) tmp_test$data.name <- tmp_nameassign(tmp_name, tmp_test)rm(list =ls(pattern ="tmp_"))}objects(pattern ="shap_")```And then the Bartlett Test of Homogeneity of Variances.```{r}#| code-fold: trueits18_div_tab_asv <- its_tab_alpha_divits18_bart_tests_asv <-c()for (i incolnames(its18_div_tab_asv[,7:ncol(its18_div_tab_asv)])) { tmp_name <- purrr::map_chr(i, ~paste0("its18_bart_asv_", .)) its18_bart_tests_asv <-append(its18_bart_tests_asv, tmp_name) tmp_test <-eval(bartlett.test(its18_div_tab_asv[[i]] ~ TEMP, data = its18_div_tab_asv)) tmp_test$data.name <- tmp_nameassign(tmp_name, tmp_test)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bart_")```Here we see which Shapiro-Wilk Normality **and** Bartlett tests were significant and which were not. So wherever the value of both p-values in `> 0.05` we can use an ANOVA, otherwise we use Kruskal-Wallis.```{r}#| echo: false#| eval: true#| warning: false#| message: falseits18_norm_res <-data.frame()for (i incolnames(its18_div_tab_asv[,7:ncol(its18_div_tab_asv)])) { tmp_get_shap <-get(purrr::map_chr(i, ~paste0("its18_shap_asv_", .))) tmp_shap_p <-round(tmp_get_shap[[2]], 4) tmp_get_bart <-get(purrr::map_chr(i, ~paste0("its18_bart_asv_", .))) tmp_bart_p <-round(tmp_get_bart[[3]], 4) tmp_test <-if(tmp_shap_p >0.05& tmp_bart_p >0.05 ){ tmp_test <-"ANOVA" } else { tmp_test <-"Kruskal-Wallis" } tmp_df <-data.frame(i, tmp_shap_p, tmp_bart_p, tmp_test) its18_norm_res <- dplyr::bind_rows(its18_norm_res, tmp_df)rm(list =ls(pattern ="tmp_"))}its18_norm_res[,1] <-gsub('_exp', '', its18_norm_res[,1])its18_norm_res <- its18_norm_res %>% tidyr::separate(col =1, into =c("metric", "dataset"), sep ="_", remove =TRUE)its18_norm_res <- its18_norm_res %>% tidyr::replace_na(list(dataset ="FULL"))its18_norm_res$dataset <- stringr::str_replace(its18_norm_res$dataset, "^fi$", "FILT") %>% stringr::str_replace(., "^pi$", "PIME") %>% stringr::str_replace(., "^pe$", "PERfect")its18_norm_res <- its18_norm_res %>% dplyr::rename(c("p-value (normality)"=3, "p-value (homogeneity)"=4, "method"=5))```<small>`r caption_tab_its("its_norm_tests")`</small>```{r}#| echo: false#| eval: trueseq_table <- its18_norm_reswrite.table(seq_table, "files/alpha/tables/its_norm_tests.txt", row.names =FALSE, sep ="\t", quote =FALSE)``````{r}#| echo: false#| eval: truereactable(seq_table,defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =0),align ="center", filterable =FALSE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold"), minWidth =100, headerVAlign ="center" ), columns =list(metric =colDef(name ="Metric", sticky ="left", style =list(borderRight ="1px solid #eee"),headerStyle =list(borderRight ="1px solid #eee"), align ="left",minWidth =100),`p-value (normality)`=colDef(name ="p-value (normality)", minWidth =100, resizable =FALSE,style =function(value) {if (value >0.05) { color <-"#2271B2" } elseif (value <0.05) { color <-"#B22271" } else { color <-"#777" }list(color = color, fontWeight ="bold") }),`p-value (homogeneity)`=colDef(name ="p-value (homogeneity)", minWidth =130, resizable =FALSE,style =function(value) {if (value >0.05) { color <-"#2271B2"#green "#", "#" } elseif (value <0.05) { color <-"#B22271"#RED } else { color <-"#777" }list(color = color, fontWeight ="bold") }),`dataset`=colDef(name ="dataset", minWidth =80, resizable =FALSE),`method`=colDef(name ="method", minWidth =120, resizable =FALSE) ), searchable =FALSE, defaultPageSize =12, showPagination =FALSE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =TRUE, showSortable =FALSE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```{{< downloadthis files/alpha/tables/its_norm_tests.txt dname="its_norm_tests" label="Download table as text file" icon="table" type="primary" >}}## Significance TestsTo begin, we need to create a hierarchy variable; a two-column matrix specifying the relationship between samples (first column) and groups (second column).```{r}#| code-fold: trueits18_hill_hier <- its_tab_alpha_divits18_hill_hier <- its18_hill_hier %>% dplyr::select("Sample_ID", "TEMP") %>% tibble::remove_rownames()its18_hill_hier <- its18_hill_hier[order(its18_hill_hier$TEMP), ]its18_hill_hier$TEMP =paste(its18_hill_hier$TEMP, 'C', sep='')its18_hill_hier <- its18_hill_hier %>% tibble::remove_rownames()saveRDS(its18_hill_hier, "files/alpha/rdata/its18_hill_hier.rds")```Again, we start by testing significance of ASV diversity for the data sets against each of the three metrics using the `div_test` function.The command is as follows:```div_test(countable = x, qvalue = i, hierarchy = hier, tree = ultrametric_tree, posthoc = TRUE)```where `x` is ASV by sample table, `i` is the q-value corresponding to the metric of interest, `hier` is the hierarchy matrix, `tree` is an ultrametric formatted phylogenetic tree (if you choose to explore lineage diversity), and `posthoc` indicates whether to run post hoc pairwise analyses.```{r}#| results: hide#| code-fold: trueqvalue <-c(0,1,2)for (i in its18_alpha_ds) {for (j in qvalue) { tmp_get <-get(purrr::map_chr(i, ~paste0(., "_tu"))) tmp_test <-div_test(tmp_get, qvalue = j,hierarchy = its18_hill_hier,posthoc =TRUE) tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt"))print(tmp_name)assign(tmp_name, tmp_test)rm(list =ls(pattern ="tmp_")) }}``````{r}#| echo: falsetmp_objects <-c("its18_ps_work", "its18_ps_filt", "its18_ps_perfect", "its18_ps_pime")tmp_metric <-data.frame(c("Observed", "Shannon exponential", "Inverse Simpson"))tmp_qvalue <-data.frame(c("0", "1", "2"))qvalue <-c(0,1,2)for (i in tmp_objects) { tmp_h_pvalue <-c()for (j in qvalue) { tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt")) tmp_get <-get(tmp_name)$homogeneity.pvalue tmp_h_pvalue <-c(append(tmp_h_pvalue, tmp_get)) } tmp_h_pvalue <-data.frame(tmp_h_pvalue) tmp_n_pvalue <-c()for (j in qvalue) { tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt")) tmp_get <-get(tmp_name)$normality.pvalue tmp_n_pvalue <-c(append(tmp_n_pvalue, tmp_get)) } tmp_n_pvalue <-data.frame(tmp_n_pvalue) tmp_method <-c()for (j in qvalue) { tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt")) tmp_get <-get(tmp_name)$method tmp_method <-c(append(tmp_method, tmp_get)) } tmp_method <-data.frame(tmp_method) tmp_phoc_method <-c()for (j in qvalue) { tmp_name <- purrr::map_chr(i, ~paste0(., "_q", j, "_adt")) tmp_get <-get(tmp_name)$posthoc.method tmp_phoc_method <-c(append(tmp_phoc_method, tmp_get)) } tmp_phoc_method <-data.frame(tmp_phoc_method) tmp_df <- dplyr::bind_cols(tmp_metric, tmp_qvalue) %>% dplyr::bind_cols(., tmp_n_pvalue) %>% dplyr::bind_cols(., tmp_h_pvalue) %>% dplyr::bind_cols(., tmp_method) %>% dplyr::bind_cols(., tmp_phoc_method) %>% dplyr::rename("metric"=1, "q-value"=2, "normality p-value"=3, "homogeneity p-value"=4, "method"=5, "posthoc method"=6) tmp_name <- purrr::map_chr(i, ~paste0(i, "_sig_tab"))assign(tmp_name, tmp_df)}``````{r}#| echo: false## Format as follows:## FALSE Kruskal-Wallis Test = its18_pime_q1_adt$test[[3]]## TRUE Tukey post-hoc = its18_ps_work_q0_adt$test[[1]][[1, 5]]############################### WORK ##################################################tmp_pvalue <-data.frame(c(its18_ps_work_q0_adt$test[[1]][[1, 5]], its18_ps_work_q1_adt$test[[1]][[1, 5]], its18_ps_work_q2_adt$test[[1]][[1, 5]]))its18_ps_work_sig_tab <- dplyr::bind_cols(its18_ps_work_sig_tab, tmp_pvalue) %>% dplyr::rename("posthoc p-value"=7)####################################Filt#############################################tmp_pvalue <-data.frame(c(its18_ps_filt_q0_adt$test[[1]][[1, 5]], its18_ps_filt_q1_adt$test[[1]][[1, 5]], its18_ps_filt_q2_adt$test[[1]][[1, 5]]))its18_ps_filt_sig_tab <- dplyr::bind_cols(its18_ps_filt_sig_tab, tmp_pvalue) %>% dplyr::rename("posthoc p-value"=7)####################################PERfect##########################################tmp_pvalue <-data.frame(c(its18_ps_perfect_q0_adt$test[[1]][[1, 5]], its18_ps_perfect_q1_adt$test[[1]][[1, 5]], its18_ps_perfect_q2_adt$test[[1]][[1, 5]]))its18_ps_perfect_sig_tab <- dplyr::bind_cols(its18_ps_perfect_sig_tab, tmp_pvalue) %>% dplyr::rename("posthoc p-value"=7)################################ PIME ################################################tmp_pvalue <-data.frame(c(its18_ps_pime_q0_adt$test[[1]][[1, 5]], its18_ps_pime_q1_adt$test[[1]][[1, 5]], its18_ps_pime_q2_adt$test[[1]][[1, 5]]))its18_ps_pime_sig_tab <- dplyr::bind_cols(its18_ps_pime_sig_tab, tmp_pvalue) %>% dplyr::rename("posthoc p-value"=7)rm(list =ls(pattern ="tmp_"))``````{r}#| echo: falseits18_ps_work_sig_tab$dataset <-"FULL"its18_ps_work_sig_tab$type <-"ASV"its18_ps_work_sig_tab$lineage <-"No"its18_ps_work_sig_tab <- its18_ps_work_sig_tab[,c(8:10,1:7)]its18_ps_pime_sig_tab$dataset <-"PIME"its18_ps_pime_sig_tab$type <-"ASV"its18_ps_pime_sig_tab$lineage <-"No"its18_ps_pime_sig_tab <- its18_ps_pime_sig_tab[,c(8:10,1:7)]its18_ps_perfect_sig_tab$dataset <-"PERfect"its18_ps_perfect_sig_tab$type <-"ASV"its18_ps_perfect_sig_tab$lineage <-"No"its18_ps_perfect_sig_tab <- its18_ps_perfect_sig_tab[,c(8:10,1:7)]its18_ps_filt_sig_tab$dataset <-"FILT"its18_ps_filt_sig_tab$type <-"ASV"its18_ps_filt_sig_tab$lineage <-"No"its18_ps_filt_sig_tab <- its18_ps_filt_sig_tab[,c(8:10,1:7)]its18_sig_tab_all <-rbind(its18_ps_work_sig_tab, its18_ps_filt_sig_tab, its18_ps_pime_sig_tab, its18_ps_perfect_sig_tab )```## Summary<small>`r caption_tab_its("its_sign_tests")`</small>```{r}#| echo: false#| eval: trueseq_table <- its18_sig_tab_allseq_table[,2:3] <-NULLseq_table <- seq_table %>% dplyr::mutate(across(.cols =c(4:5), round, digits =4)) write.table(seq_table, "files/alpha/tables/its_sign_tests.txt", row.names =FALSE, sep ="\t", quote =FALSE)``````{r}#| echo: false#| eval: truereactable(seq_table,defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =0),align ="center", filterable =FALSE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold"), minWidth =80 ), columns =list(dataset =colDef(name ="Dataset", sticky ="left", style =list(borderRight ="1px solid #eee"),headerStyle =list(borderRight ="1px solid #eee"), align ="left",minWidth =100),`posthoc p-value`=colDef(name ="posthoc p-value", style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color, fontWeight ="bold") }, cell =function(value) format(value, digits =3, scientific =TRUE) ),metric =colDef(name ="metric", minWidth =140, align ="left"),`q-value`=colDef(name ="q-value", minWidth =70, align ="center"),`normality p-value`=colDef(name ="p-value (normality)", minWidth =100,),`homogeneity p-value`=colDef(name ="p-value (homogeneity)", minWidth =130),`dataset`=colDef(name ="dataset", minWidth =80),`method`=colDef(name ="method", minWidth =120) ), searchable =FALSE, defaultPageSize =12, showPagination =FALSE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =FALSE, showSortable =FALSE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.7em")))rm(seq_table)```{{< downloadthis files/alpha/tables/its_sign_tests.txt dname="its_sign_tests" label="Download table as text file" icon="table" type="primary" >}}## PostHoc Analyses First let's check the results of each posthoc analysis.::: {.column-body}::: {.panel-tabset}### Observed (q-value = 0)```{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvf0_lab <-"FULL (Observed)"its18_asvf0_labits18_ps_work_q0_adt$posthoc.methoddata.frame(its18_ps_work_q0_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvl0_lab <-"FILT (Observed)"its18_asvl0_labits18_ps_filt_q0_adt$posthoc.methoddata.frame(its18_ps_filt_q0_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvp0_lab <-"PIME (Observed)"its18_asvp0_labits18_ps_pime_q0_adt$posthoc.methoddata.frame(its18_ps_pime_q0_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvr0_lab <-"PERfect (Observed)"its18_asvr0_labits18_ps_perfect_q0_adt$posthoc.methoddata.frame(its18_ps_perfect_q0_adt$posthoc)```### Shannon exp (q-value = 1)```{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvf1_lab <-"FULL (Shannon exponential)"its18_asvf1_labits18_ps_work_q1_adt$posthoc.methoddata.frame(its18_ps_work_q1_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvl1_lab <-"FILT (Shannon exponential)"its18_asvl1_labits18_ps_filt_q1_adt$posthoc.methoddata.frame(its18_ps_filt_q1_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvp1_lab <-"PIME (Shannon exponential)"its18_asvp1_labits18_ps_pime_q1_adt$posthoc.methoddata.frame(its18_ps_pime_q1_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvr1_lab <-"PERfect (Shannon exponential)"its18_asvr1_labits18_ps_perfect_q1_adt$posthoc.methoddata.frame(its18_ps_perfect_q1_adt$posthoc)```### Inverse Simpson (q-value = 2)```{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvf2_lab <-"FULL (Inverse Simpson)"its18_asvf2_labits18_ps_work_q2_adt$posthoc.methoddata.frame(its18_ps_work_q2_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvl2_lab <-"FILT (Inverse Simpson)"its18_asvl2_labits18_ps_filt_q2_adt$posthoc.methoddata.frame(its18_ps_filt_q2_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvp2_lab <-"PIME (Inverse Simpson)"its18_asvp2_labits18_ps_pime_q2_adt$posthoc.methoddata.frame(its18_ps_pime_q2_adt$posthoc)``````{r}#| echo: false#| eval: true#| results: hold#| comment: '' its18_asvr2_lab <-"PERfect (Inverse Simpson)"its18_asvr2_labits18_ps_perfect_q2_adt$posthoc.methoddata.frame(its18_ps_perfect_q2_adt$posthoc)```::::::Now we can plot the results from the posthoc analyses for each metric and data set using the function `div_test_plot_jjs`. I modified the original function (`div_test_plot`) to control a little of the formatting.The command is as follows:```div_test_plot(divtest = x, chart = "type", colour = col.pal, posthoc = TRUE, threshold = value))```where `x` is the results from the `div_test` function, `"type"` is chart type (box, jitter, or violin), `colour` is is a color palette, `posthoc` indicates whether to run posthoc pairwise analyses, and `value` is the maximum p-value to show in pairwise posthoc results. **WARNING** if none of the posthoc results are below the specified threshold, the function will throw an error. Therefore, until this is fixed, all posthoc values are shown.```{r}#| code-fold: truesource("hack_code/div_test_plot_jjs.R")rm(list =ls(pattern ="_adt_plot"))for (i inobjects(pattern ="_adt")) { tmp_name <- purrr::map_chr(i, ~paste0(., "_plot")) tmp_get <-get(i) tmp_df <-div_test_plot_jjs(tmp_get, chart ="box",colour = swel_col, posthoc =TRUE) tmp_df <-ggpar(tmp_df, legend ="none")print(tmp_name)assign(tmp_name, tmp_df)rm(list =ls(pattern ="tmp_"))}its18_ps_work_q0_adt_plot``````{r}#| echo: false#| eval: falseits18_ps_work_q0_adt_plot <- its18_ps_work_q0_adt_plot +theme(axis.title.x =element_blank()) +ggtitle(its18_asvf0_lab) +theme(plot.title =element_text(size =20, face ="bold"))its18_ps_filt_q0_adt_plot <- its18_ps_filt_q0_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(its18_asvl0_lab) +theme(plot.title =element_text(size =20, face ="bold"))its18_ps_perfect_q0_adt_plot <- its18_ps_perfect_q0_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(its18_asvr0_lab) +theme(plot.title =element_text(size =20, face ="bold"))its18_ps_pime_q0_adt_plot <- its18_ps_pime_q0_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(its18_asvp0_lab)+theme(plot.title =element_text(size =20, face ="bold"))#####################its18_ps_work_q1_adt_plot <- its18_ps_work_q1_adt_plot +theme(axis.title.x =element_blank()) +ggtitle(its18_asvf1_lab)+theme(plot.title =element_text(size =20, face ="bold"))its18_ps_filt_q1_adt_plot <- its18_ps_filt_q1_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(its18_asvl1_lab) +theme(plot.title =element_text(size =20, face ="bold"))its18_ps_perfect_q1_adt_plot <- its18_ps_perfect_q1_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(its18_asvr1_lab) +theme(plot.title =element_text(size =20, face ="bold"))its18_ps_pime_q1_adt_plot <- its18_ps_pime_q1_adt_plot +theme(axis.title.y =element_blank(),axis.title.x =element_blank()) +ggtitle(its18_asvp1_lab) +theme(plot.title =element_text(size =20, face ="bold"))#####################its18_ps_work_q2_adt_plot <- its18_ps_work_q2_adt_plot +ggtitle(its18_asvf2_lab) +theme(plot.title =element_text(size =20, face ="bold"))its18_ps_filt_q2_adt_plot <- its18_ps_filt_q2_adt_plot +theme(axis.title.y =element_blank()) +ggtitle(its18_asvl2_lab) +theme(plot.title =element_text(size =20, face ="bold"))its18_ps_perfect_q2_adt_plot <- its18_ps_perfect_q2_adt_plot +theme(axis.title.y =element_blank()) +ggtitle(its18_asvr2_lab) +theme(plot.title =element_text(size =20, face ="bold"))its18_ps_pime_q2_adt_plot <- its18_ps_pime_q2_adt_plot +theme(axis.title.y =element_blank()) +ggtitle(its18_asvp2_lab) +theme(plot.title =element_text(size =20, face ="bold"))its18_alph_div_plots_asv <-ggarrange( its18_ps_work_q0_adt_plot, its18_ps_filt_q0_adt_plot, its18_ps_perfect_q0_adt_plot, its18_ps_pime_q0_adt_plot, its18_ps_work_q1_adt_plot, its18_ps_filt_q1_adt_plot, its18_ps_perfect_q1_adt_plot, its18_ps_pime_q1_adt_plot, its18_ps_work_q2_adt_plot, its18_ps_filt_q2_adt_plot, its18_ps_perfect_q2_adt_plot, its18_ps_pime_q2_adt_plot, ncol=4, nrow =3)its18_alph_div_plots_asvdev.off()png("files/alpha/figures/its18_alph_div_plots_asv.png",height=32, width=60, units ='cm', res =600, bg ="white")its18_alph_div_plots_asvdev.off()pdf("files/alpha/figures/its18_alph_div_plots_asv.pdf",height =10, width =11)its18_alph_div_plots_asvdev.off()system("cp files/alpha/figures/its18_alph_div_plots_asv.png include/alpha/its18_alph_div_plots_asv.png")```## Alpha Diversity PlotsPosthoc adjusted p-values given for each pairwise comparison.```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 5#| column: body-outsetknitr::include_graphics("include/alpha/its18_alph_div_plots_asv.png")```<small>`r caption_fig_its("its_alpha_div_plots")`</small>```{r}#| echo: falsesave.image("page_build/alpha_wf.rdata")``````{r}#| echo: falsesamp_ps <-c("its18_ps_work", "its18_ps_filt", "its18_ps_pime", "its18_ps_perfect")for (i in samp_ps) {dir.create(paste("files/alpha/figures/its/", i, sep =""), recursive =TRUE) tmp_get_q0 <-get(paste0(i, "_q0_adt_plot", sep ="")) tmp_get_q1 <-get(paste0(i, "_q1_adt_plot", sep ="")) tmp_get_q2 <-get(paste0(i, "_q2_adt_plot", sep ="")) ggplot2::ggsave(tmp_get_q0, file =paste0("files/alpha/figures/its/", i, "/", i, "_q0_adt_plot", ".png", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q0, file =paste0("files/alpha/figures/its/", i, "/", i, "_q0_adt_plot", ".pdf", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q1, file =paste0("files/alpha/figures/its/", i, "/", i, "_q1_adt_plot", ".png", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q1, file =paste0("files/alpha/figures/its/", i, "/", i, "_q1_adt_plot", ".pdf", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q2, file =paste0("files/alpha/figures/its/", i, "/", i, "_q2_adt_plot", ".png", sep =""), width =5, height =3) ggplot2::ggsave(tmp_get_q2, file =paste0("files/alpha/figures/its/", i, "/", i, "_q2_adt_plot", ".pdf", sep =""), width =5, height =3)rm(list =ls(pattern ="tmp_"))} ``````{r}#| include: false#| eval: trueremove(list =ls())```# Workflow Output Data products generated in this workflow can be downloaded from figshare.<iframe src="https://widgets.figshare.com/articles/16826779/embed?show_title=1" width="100%" height="251" allowfullscreen frameborder="0"></iframe></br><a href="beta.html" class="btnnav button_round" style="float: right;">Next workflow:<br> 6. Beta Diversity & Dispersion Estimates</a><a href="taxa.html" class="btnnav button_round" style="float: left;">Previous workflow:<br> 4. Taxonomic Diversity</a><br><br>```{r}#| message: false #| results: hide#| eval: true#| echo: falseremove(list =ls())### COmmon formatting scripts### NOTE: captioner.R must be read BEFORE captions_XYZ.Rsource(file.path("assets", "functions.R"))```#### Source Code {.appendix}The source code for this page can be accessed on GitHub `r fa(name = "github")` by [clicking this link](`r source_code()`). #### Data Availability {.appendix}Data generated in this workflow and the Rdata need to run the workflow can be accessed on figshare at [10.25573/data.16826779](https://doi.org/10.25573/data.16826779).#### Last updated on {.appendix}```{r}#| echo: false#| eval: trueSys.time()```