In this workflow we compare the environmental metadata with the microbial community data. Part 1 involves the 16S rRNA community data and Part 2 deals with the ITS community data. Each workflow contains the same major steps:
Metadata Normality Tests: Shapiro-Wilk Normality Test to test whether each matadata parameter is normally distributed.
Normalize Parameters: R package bestNormalize to find and execute the best normalizing transformation.
Split Metadata parameters into groups: a) Environmental and edaphic properties, b) Microbial functional responses, and c) Temperature adaptation properties.
Autocorrelation Tests: Test all possible pair-wise comparisons, on both normalized and non-normalized data sets, for each group.
Remove autocorrelated parameters from each group.
Dissimilarity Correlation Tests: Use Mantel Tests to see if any on the metadata groups are significantly correlated with the community data.
Best Subset of Variables: Determine which of the metadata parameters from each group are the most strongly correlated with the community data. For this we use the bioenv function from the vegan(Oksanen et al. 2012) package.
Distance-based Redundancy Analysis: Ordination analysis of samples and metadata vector overlays using capscale, also from the vegan package.
Workflow Input
Files needed to run this workflow can be downloaded from figshare. You will also need the metadata table, which can be accessed below.
Metadata
In this section we provide access to the complete environmental metadata containing 61 parameters across all 15 samples.
(common) Table 1 | Metadata values for each sample.
Multivariate Summary
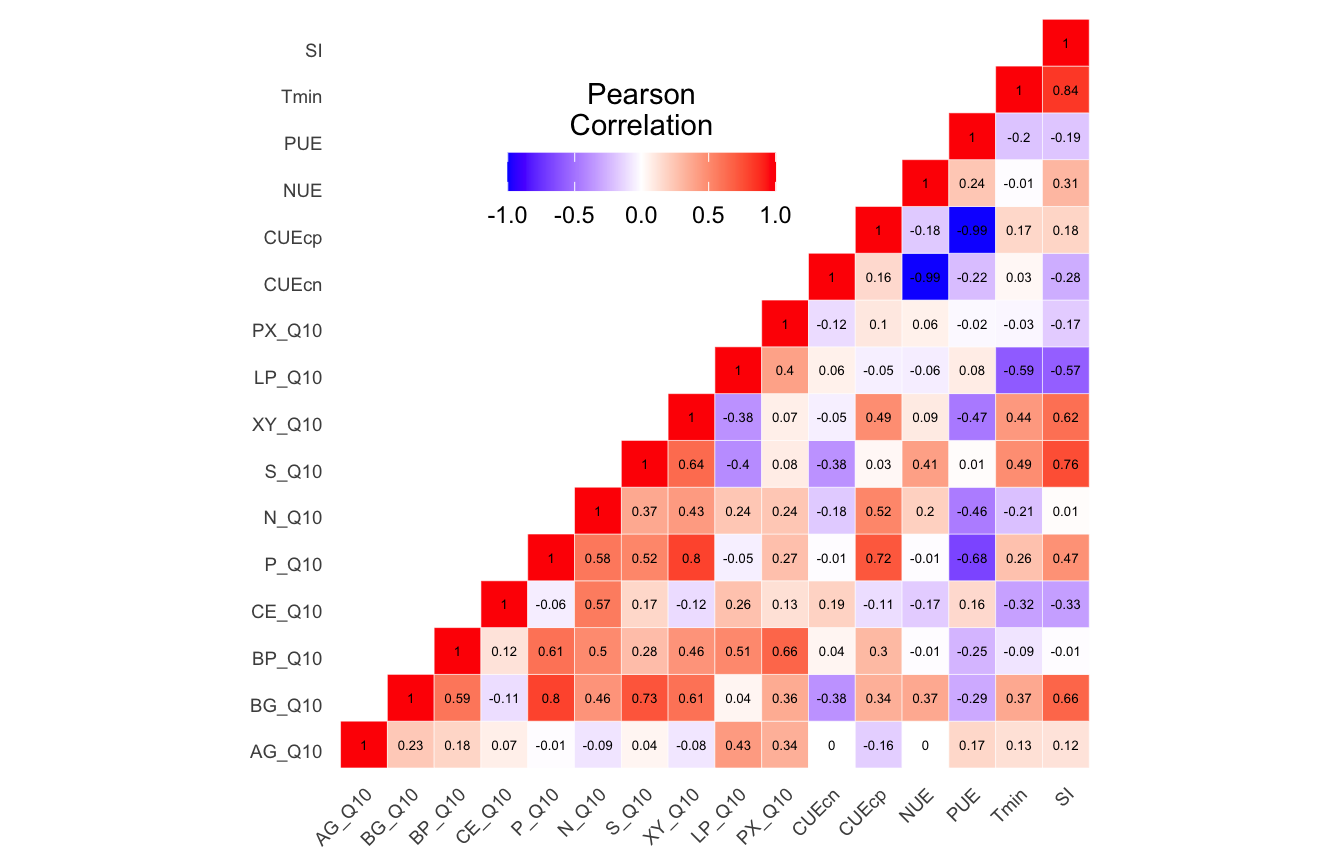
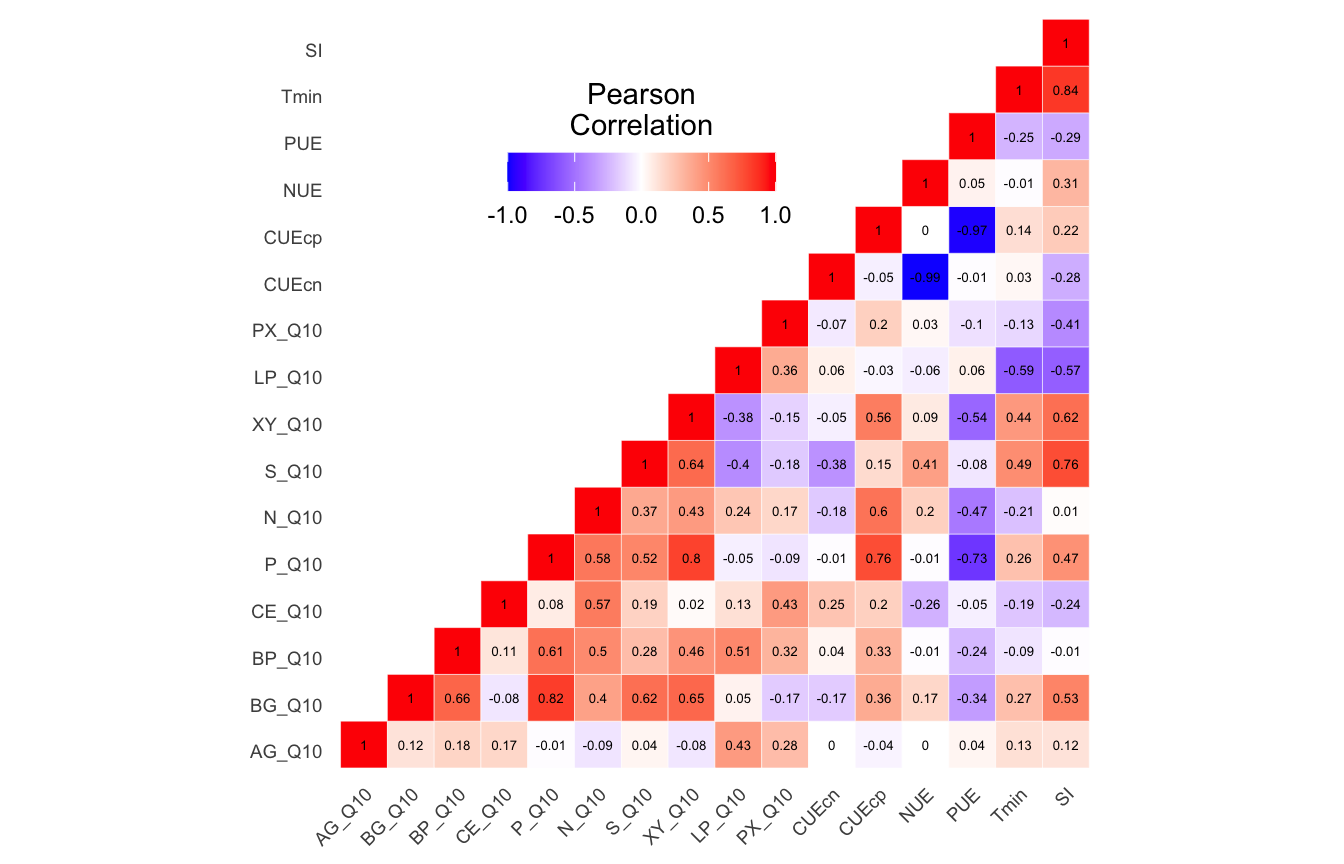
Summary results for of envfit and bioenv tests for edaphic properties, soil functional response, and temperature adaptation for both 16S rRNA and ITS data sets.
16S rRNA
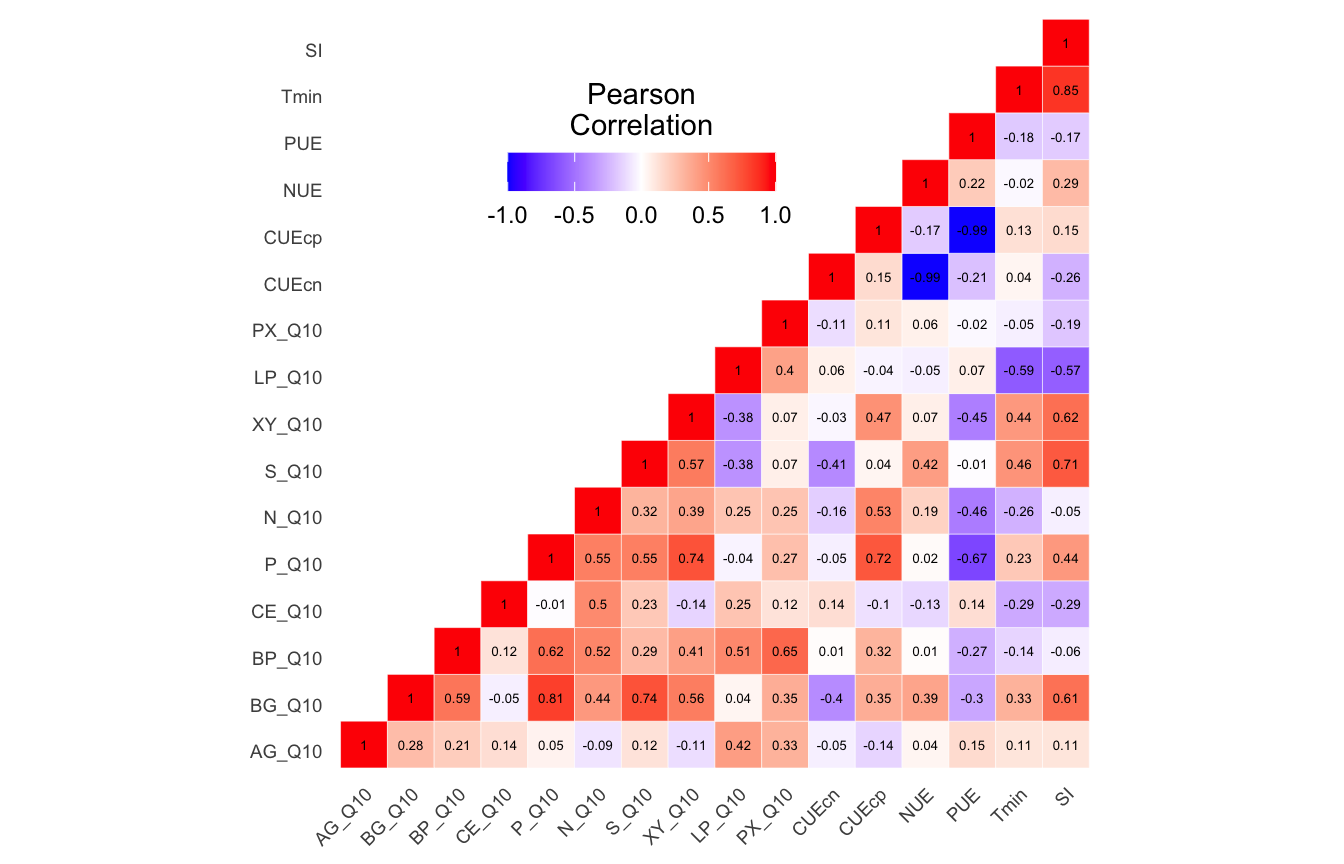
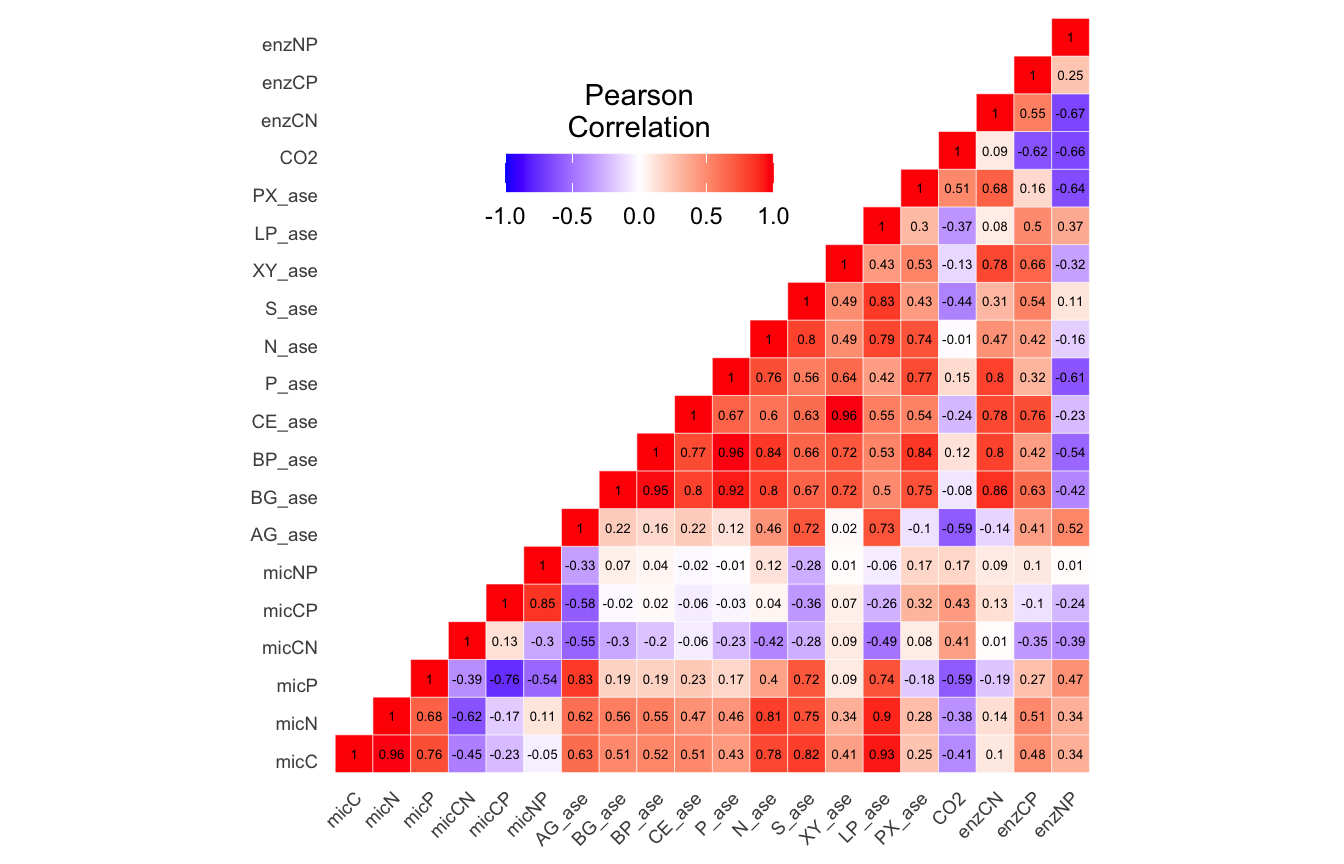
Metadata parameters removed based on autocorrelation results.
Before proceeding, we need to test each parameter in the metadata to see which ones are and are not normally distributed. For that, we use the Shapiro-Wilk Normality Test. Here we only need one of the metadata files.
Show the results of each normality test for metadata parameters
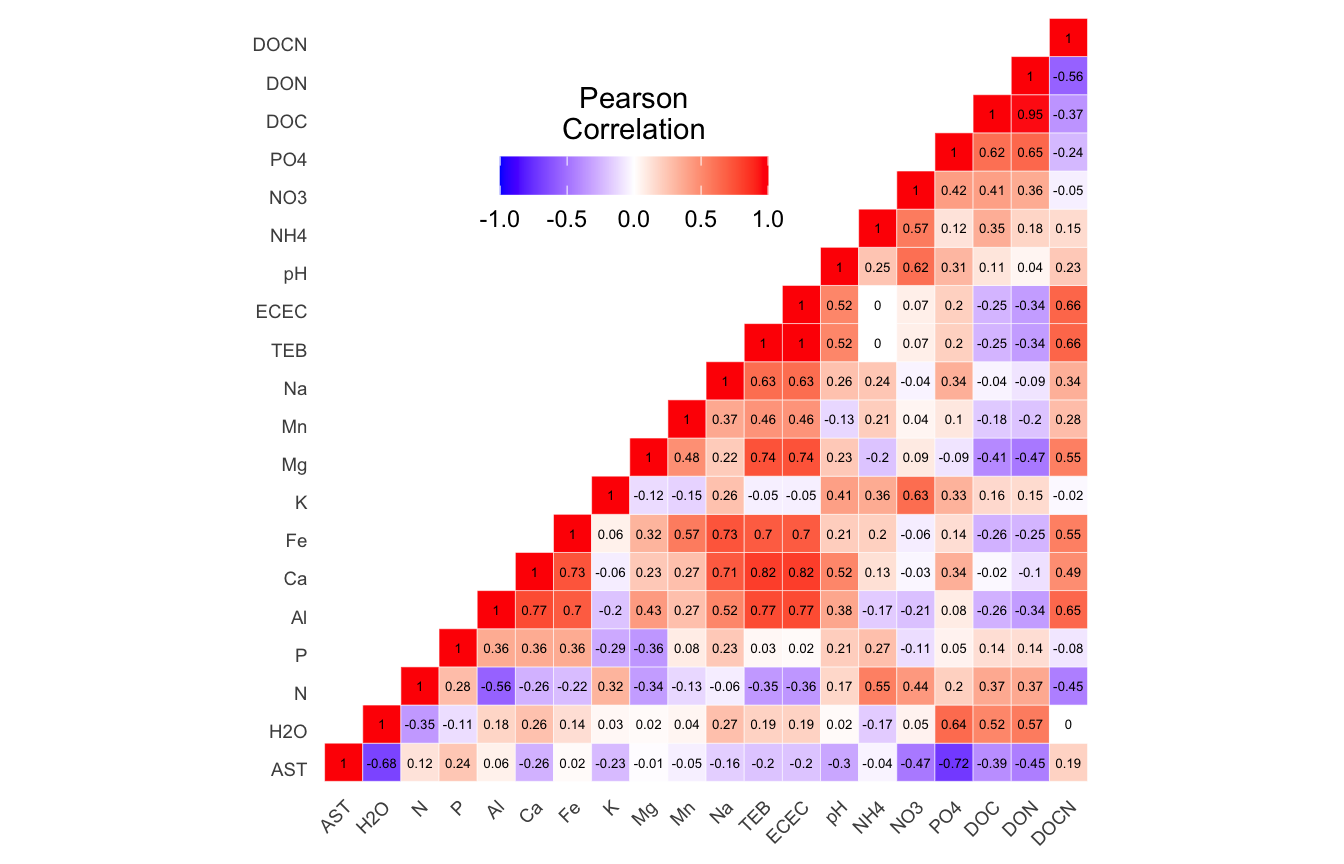
(16S rRNA) Table 2 | Results of the Shapiro-Wilk Normality Tests. P-values in red are significance (p-value < 0.05) meaning the parameter needs to be normalized.
Looks like we need to transform 25 metadata parameters.
Normalize Parameters
Here we use the R package bestNormalize to find and execute the best normalizing transformation. The function will test the following normalizing transformations:
arcsinh_x performs an arcsinh transformation.
boxcox Perform a Box-Cox transformation and center/scale a vector to attempt normalization. boxcox estimates the optimal value of lambda for the Box-Cox transformation. The function will return an error if a user attempt to transform nonpositive data.
yeojohnson Perform a Yeo-Johnson Transformation and center/scale a vector to attempt normalization. yeojohnson estimates the optimal value of lambda for the Yeo-Johnson transformation. The Yeo-Johnson is similar to the Box-Cox method, however it allows for the transformation of nonpositive data as well.
orderNorm The Ordered Quantile (ORQ) normalization transformation, orderNorm(), is a rank-based procedure by which the values of a vector are mapped to their percentile, which is then mapped to the same percentile of the normal distribution. Without the presence of ties, this essentially guarantees that the transformation leads to a uniform distribution.
log_x performs a simple log transformation. The parameter a is essentially estimated by the training set by default (estimated as the minimum possible to some extent epsilon), while the base must be specified beforehand. The default base of the log is 10.
sqrt_x performs a simple square-root transformation. The parameter a is essentially estimated by the training set by default (estimated as the minimum possible), while the base must be specified beforehand.
exp_x performs a simple exponential transformation.
See this GitHub issue (#5) for a description on getting reproducible results. Apparently, you can get different results because the bestNormalize() function uses repeated cross-validation (and doesn’t automatically set the seed), so the results will be slightly different each time the function is executed.
Show the code
set.seed(119)for(iinmd_to_tranform){tmp_md<-ssu18_select_mc$map_loadedtmp_best_norm<-bestNormalize(tmp_md[[i]], r =1, k =5, loo =TRUE)tmp_name<-purrr::map_chr(i, ~paste0(., "_best_norm_test"))assign(tmp_name, tmp_best_norm)print(tmp_name)rm(list =ls(pattern ="tmp_"))}
Show the chosen transformations
## bestNormalize Chosen transformation of AST ##
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
25.800 26.275 28.800 30.280 41.770
_____________________________________
## bestNormalize Chosen transformation of H2O ##
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
0.204 0.308 0.370 0.384 0.401
_____________________________________
## bestNormalize Chosen transformation of Al ##
Standardized asinh(x) Transformation with 15 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 0.0059997
- sd (before standardization) = 0.008280287
_____________________________________
## bestNormalize Chosen transformation of Ca ##
orderNorm Transformation with 15 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
25.49 27.19 29.23 34.36 39.79
_____________________________________
## bestNormalize Chosen transformation of Fe ##
Standardized asinh(x) Transformation with 15 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 0.0119984
- sd (before standardization) = 0.01264569
_____________________________________
## bestNormalize Chosen transformation of TEB ##
orderNorm Transformation with 15 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
41.6 43.3 45.1 51.4 61.7
_____________________________________
## bestNormalize Chosen transformation of ECEC ##
orderNorm Transformation with 15 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
41.7 43.3 45.3 51.5 61.8
_____________________________________
## bestNormalize Chosen transformation of minNO3 ##
Standardized Box Cox Transformation with 15 nonmissing obs.:
Estimated statistics:
- lambda = -0.3344538
- mean (before standardization) = 1.907818
- sd (before standardization) = 0.2388776
_____________________________________
## bestNormalize Chosen transformation of minTIN ##
orderNorm Transformation with 15 nonmissing obs and ties
- 14 unique values
- Original quantiles:
0% 25% 50% 75% 100%
8.760 14.995 23.080 39.935 110.750
_____________________________________
## bestNormalize Chosen transformation of DOC ##
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
30.800 44.070 56.680 64.385 118.740
_____________________________________
## bestNormalize Chosen transformation of DOCN ##
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
9.440 10.290 11.150 12.095 19.270
_____________________________________
## bestNormalize Chosen transformation of micCN ##
orderNorm Transformation with 15 nonmissing obs and ties
- 14 unique values
- Original quantiles:
0% 25% 50% 75% 100%
4.420 4.585 4.720 5.415 5.850
_____________________________________
## bestNormalize Chosen transformation of BG_ase ##
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
3.010 3.735 4.220 5.795 10.480
_____________________________________
## bestNormalize Chosen transformation of BP_ase ##
Standardized Log_b(x + a) Transformation with 15 nonmissing obs.:
Relevant statistics:
- a = 0
- b = 10
- mean (before standardization) = 0.5217757
- sd (before standardization) = 0.2001498
_____________________________________
## bestNormalize Chosen transformation of CE_ase ##
Standardized Log_b(x + a) Transformation with 15 nonmissing obs.:
Relevant statistics:
- a = 0
- b = 10
- mean (before standardization) = 0.03306978
- sd (before standardization) = 0.1652591
_____________________________________
## bestNormalize Chosen transformation of P_ase ##
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
13.020 15.130 19.210 24.455 44.040
_____________________________________
## bestNormalize Chosen transformation of N_ase ##
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
2.57 2.93 3.58 5.04 9.43
_____________________________________
## bestNormalize Chosen transformation of XY_ase ##
orderNorm Transformation with 15 nonmissing obs and ties
- 13 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.66 0.81 1.11 1.39 2.52
_____________________________________
## bestNormalize Chosen transformation of BG_Q10 ##
orderNorm Transformation with 15 nonmissing obs and ties
- 13 unique values
- Original quantiles:
0% 25% 50% 75% 100%
1.310 1.360 1.390 1.545 1.760
_____________________________________
## bestNormalize Chosen transformation of BP_Q10 ##
orderNorm Transformation with 15 nonmissing obs and ties
- 11 unique values
- Original quantiles:
0% 25% 50% 75% 100%
1.340 1.415 1.440 1.530 1.740
_____________________________________
## bestNormalize Chosen transformation of CO2 ##
orderNorm Transformation with 15 nonmissing obs and ties
- 14 unique values
- Original quantiles:
0% 25% 50% 75% 100%
2.080 4.370 5.270 10.585 40.470
_____________________________________
## bestNormalize Chosen transformation of PX_ase ##
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
81.250 95.565 119.750 218.370 339.600
_____________________________________
## bestNormalize Chosen transformation of PX_Q10 ##
Standardized Yeo-Johnson Transformation with 15 nonmissing obs.:
Estimated statistics:
- lambda = -4.999946
- mean (before standardization) = 0.1968247
- sd (before standardization) = 0.001294011
_____________________________________
## bestNormalize Chosen transformation of CUEcp ##
Standardized Box Cox Transformation with 15 nonmissing obs.:
Estimated statistics:
- lambda = -0.9999576
- mean (before standardization) = -4.229896
- sd (before standardization) = 0.9115063
_____________________________________
## bestNormalize Chosen transformation of PUE ##
orderNorm Transformation with 15 nonmissing obs and ties
- 7 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.770 0.880 0.900 0.905 0.920
_____________________________________
Show the complete bestNormalize results
## Results of bestNormalize for AST ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 3.4
- Box-Cox: 1.8
- Center+scale: 4.2
- Exp(x): 21.2667
- Log_b(x+a): 3.4
- orderNorm (ORQ): 0.2
- sqrt(x + a): 4.2
- Yeo-Johnson: 2.8667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
25.800 26.275 28.800 30.280 41.770
_____________________________________
## Results of bestNormalize for H2O ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 6.8667
- Box-Cox: 5.8
- Center+scale: 5.8
- Exp(x): 5.8
- Log_b(x+a): 6.0667
- orderNorm (ORQ): 0.7333
- sqrt(x + a): 6.8667
- Yeo-Johnson: 5.8
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
0.204 0.308 0.370 0.384 0.401
_____________________________________
## Results of bestNormalize for Al ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 8.2
- Center+scale: 8.2
- Exp(x): 8.2
- Log_b(x+a): 8.2
- orderNorm (ORQ): 8.2
- sqrt(x + a): 8.2
- Yeo-Johnson: 8.2
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized asinh(x) Transformation with 15 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 0.0059997
- sd (before standardization) = 0.008280287
_____________________________________
## Results of bestNormalize for Ca ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.8
- Box-Cox: 1.8
- Center+scale: 3.4
- Exp(x): 18.0667
- Log_b(x+a): 1.8
- orderNorm (ORQ): 0.2
- sqrt(x + a): 1.8
- Yeo-Johnson: 1.2667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
25.49 27.19 29.23 34.36 39.79
_____________________________________
## Results of bestNormalize for Fe ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 6.3333
- Center+scale: 6.3333
- Exp(x): 6.3333
- Log_b(x+a): 6.3333
- orderNorm (ORQ): 6.3333
- sqrt(x + a): 6.3333
- Yeo-Johnson: 6.3333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized asinh(x) Transformation with 15 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 0.0119984
- sd (before standardization) = 0.01264569
_____________________________________
## Results of bestNormalize for TEB ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 2.3333
- Box-Cox: 1.2667
- Center+scale: 6.0667
- Exp(x): 18.0667
- Log_b(x+a): 2.3333
- orderNorm (ORQ): 0.4667
- sqrt(x + a): 5.5333
- Yeo-Johnson: 1.5333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
41.6 43.3 45.1 51.4 61.7
_____________________________________
## Results of bestNormalize for ECEC ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.2667
- Box-Cox: 1.2667
- Center+scale: 6.0667
- Exp(x): 18.0667
- Log_b(x+a): 1.2667
- orderNorm (ORQ): 0.4667
- sqrt(x + a): 3.9333
- Yeo-Johnson: 1.5333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
41.7 43.3 45.3 51.5 61.8
_____________________________________
## Results of bestNormalize for minNO3 ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.8
- Box-Cox: 1.2667
- Center+scale: 3.9333
- Exp(x): 21.2667
- Log_b(x+a): 1.8
- orderNorm (ORQ): 1.2667
- sqrt(x + a): 1.8
- Yeo-Johnson: 1.2667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized Box Cox Transformation with 15 nonmissing obs.:
Estimated statistics:
- lambda = -0.3344538
- mean (before standardization) = 1.907818
- sd (before standardization) = 0.2388776
_____________________________________
## Results of bestNormalize for minTIN ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.8
- Box-Cox: 1.8
- Center+scale: 3.9333
- Exp(x): 21.2667
- Log_b(x+a): 1.8
- orderNorm (ORQ): 0.7333
- sqrt(x + a): 1.8
- Yeo-Johnson: 1.8
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 14 unique values
- Original quantiles:
0% 25% 50% 75% 100%
8.760 14.995 23.080 39.935 110.750
_____________________________________
## Results of bestNormalize for DOC ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 2.3333
- Box-Cox: 1.2667
- Center+scale: 2.8667
- Exp(x): 21.2667
- Log_b(x+a): 2.3333
- orderNorm (ORQ): 0.4667
- sqrt(x + a): 2.8667
- Yeo-Johnson: 1.2667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
30.800 44.070 56.680 64.385 118.740
_____________________________________
## Results of bestNormalize for DOCN ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 2.3333
- Box-Cox: 0.7333
- Center+scale: 3.9333
- Exp(x): 21.2667
- Log_b(x+a): 2.3333
- orderNorm (ORQ): 0.2
- sqrt(x + a): 3.1333
- Yeo-Johnson: 0.2
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
9.440 10.290 11.150 12.095 19.270
_____________________________________
## Results of bestNormalize for micCN ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 3.1333
- Box-Cox: 3.1333
- Center+scale: 3.1333
- Exp(x): 5.5333
- Log_b(x+a): 3.1333
- orderNorm (ORQ): 0.2
- sqrt(x + a): 3.1333
- Yeo-Johnson: 3.1333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 14 unique values
- Original quantiles:
0% 25% 50% 75% 100%
4.420 4.585 4.720 5.415 5.850
_____________________________________
## Results of bestNormalize for BG_ase ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 2.8667
- Box-Cox: 2.3333
- Center+scale: 5
- Exp(x): 21.2667
- Log_b(x+a): 2.8667
- orderNorm (ORQ): 0.4667
- sqrt(x + a): 2.8667
- Yeo-Johnson: 2.3333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
3.010 3.735 4.220 5.795 10.480
_____________________________________
## Results of bestNormalize for BP_ase ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.5333
- Box-Cox: 1.5333
- Center+scale: 3.1333
- Exp(x): 11.9333
- Log_b(x+a): 0.7333
- orderNorm (ORQ): 0.7333
- sqrt(x + a): 3.1333
- Yeo-Johnson: 1.5333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized Log_b(x + a) Transformation with 15 nonmissing obs.:
Relevant statistics:
- a = 0
- b = 10
- mean (before standardization) = 0.5217757
- sd (before standardization) = 0.2001498
_____________________________________
## Results of bestNormalize for CE_ase ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1
- Box-Cox: 0.7333
- Center+scale: 1.8
- Exp(x): 4.2
- Log_b(x+a): 0.4667
- orderNorm (ORQ): 1
- sqrt(x + a): 1
- Yeo-Johnson: 0.4667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized Log_b(x + a) Transformation with 15 nonmissing obs.:
Relevant statistics:
- a = 0
- b = 10
- mean (before standardization) = 0.03306978
- sd (before standardization) = 0.1652591
_____________________________________
## Results of bestNormalize for P_ase ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 2.0667
- Box-Cox: 1.2667
- Center+scale: 3.6667
- Exp(x): 21.2667
- Log_b(x+a): 2.0667
- orderNorm (ORQ): 0.2
- sqrt(x + a): 2.3333
- Yeo-Johnson: 1.2667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
13.020 15.130 19.210 24.455 44.040
_____________________________________
## Results of bestNormalize for N_ase ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.2667
- Box-Cox: 1.8
- Center+scale: 3.1333
- Exp(x): 17.8
- Log_b(x+a): 1.2667
- orderNorm (ORQ): 0.2
- sqrt(x + a): 3.1333
- Yeo-Johnson: 1.2667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
2.57 2.93 3.58 5.04 9.43
_____________________________________
## Results of bestNormalize for XY_ase ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 0.7333
- Box-Cox: 0.7333
- Center+scale: 2.8667
- Exp(x): 6.3333
- Log_b(x+a): 0.7333
- orderNorm (ORQ): 0.2
- sqrt(x + a): 1.8
- Yeo-Johnson: 0.7333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 13 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.66 0.81 1.11 1.39 2.52
_____________________________________
## Results of bestNormalize for BG_Q10 ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 2.3333
- Box-Cox: 2.3333
- Center+scale: 2.3333
- Exp(x): 6.0667
- Log_b(x+a): 2.3333
- orderNorm (ORQ): 0.2
- sqrt(x + a): 2.3333
- Yeo-Johnson: 2.3333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 13 unique values
- Original quantiles:
0% 25% 50% 75% 100%
1.310 1.360 1.390 1.545 1.760
_____________________________________
## Results of bestNormalize for BP_Q10 ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 2.6
- Box-Cox: 2.6
- Center+scale: 2.6
- Exp(x): 2.6
- Log_b(x+a): 2.6
- orderNorm (ORQ): 0.7333
- sqrt(x + a): 2.6
- Yeo-Johnson: 2.3333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 11 unique values
- Original quantiles:
0% 25% 50% 75% 100%
1.340 1.415 1.440 1.530 1.740
_____________________________________
## Results of bestNormalize for CO2 ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.5333
- Box-Cox: 2.6
- Center+scale: 11.9333
- Exp(x): 21.2667
- Log_b(x+a): 1.5333
- orderNorm (ORQ): 1.2667
- sqrt(x + a): 3.4
- Yeo-Johnson: 2.0667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 14 unique values
- Original quantiles:
0% 25% 50% 75% 100%
2.080 4.370 5.270 10.585 40.470
_____________________________________
## Results of bestNormalize for PX_ase ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 2.3333
- Box-Cox: 1.2667
- Center+scale: 7.6667
- Exp(x): 21.2667
- Log_b(x+a): 2.3333
- orderNorm (ORQ): 0.2
- sqrt(x + a): 2.8667
- Yeo-Johnson: 1.2667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
81.250 95.565 119.750 218.370 339.600
_____________________________________
## Results of bestNormalize for PX_Q10 ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 14.6
- Box-Cox: 4.2
- Center+scale: 21.2667
- Exp(x): 21.2667
- Log_b(x+a): 11.9333
- orderNorm (ORQ): 2.0667
- sqrt(x + a): 21.2667
- Yeo-Johnson: 1.5333
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized Yeo-Johnson Transformation with 15 nonmissing obs.:
Estimated statistics:
- lambda = -4.999946
- mean (before standardization) = 0.1968247
- sd (before standardization) = 0.001294011
_____________________________________
## Results of bestNormalize for CUEcp ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 3.9333
- Box-Cox: 1.5333
- Center+scale: 3.9333
- Exp(x): 3.9333
- Log_b(x+a): 2.0667
- orderNorm (ORQ): 1.5333
- sqrt(x + a): 2.0667
- Yeo-Johnson: 2.0667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized Box Cox Transformation with 15 nonmissing obs.:
Estimated statistics:
- lambda = -0.9999576
- mean (before standardization) = -4.229896
- sd (before standardization) = 0.9115063
_____________________________________
## Results of bestNormalize for PUE ##
Best Normalizing transformation with 15 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 8.2
- Box-Cox: 9.8
- Center+scale: 8.2
- Exp(x): 3.9333
- Log_b(x+a): 9.8
- orderNorm (ORQ): 2.0667
- sqrt(x + a): 9.8
- Yeo-Johnson: 9.8
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 15 nonmissing obs and ties
- 7 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.770 0.880 0.900 0.905 0.920
_____________________________________
Great, now we can add the normalized transformed data back to our mctoolsr metadata file.
Ok. Looks like bestNormalize was unable to find a suitable transformation for Al and Fe. This is likely because there is very little variation in these metadata and/or there are too few significant digits.
Normalized Metadata
Finally, here is a new summary table that includes all of the normalized data.
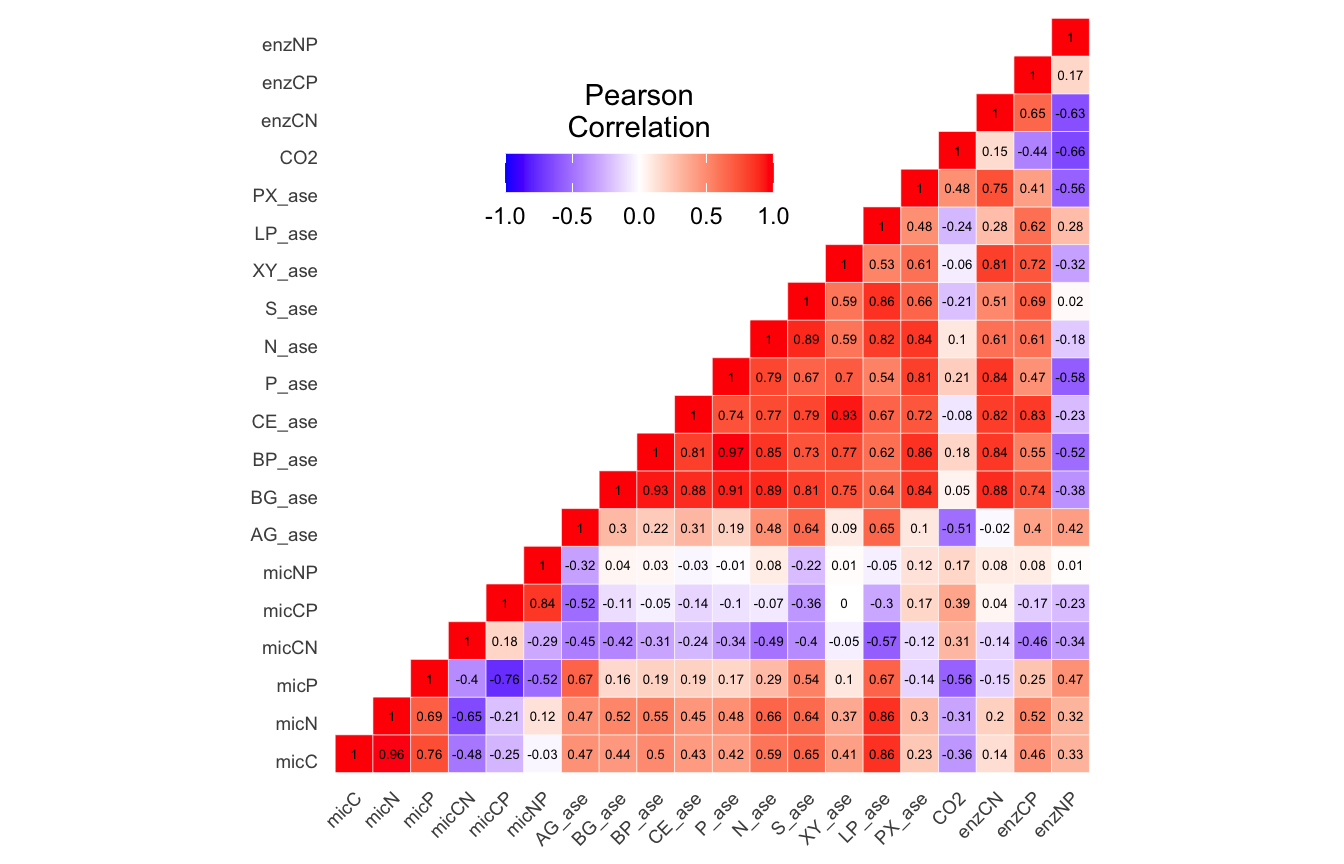
(16S rRNA) Table 3 | Results of bestNormalize function applied to each parameter.
Autocorrelation Tests
Next, we test the metadata for autocorrelations. Do we do this on the original data or the transformed data? No idea, so let’s do both.
Let’s see if any on the metadata groups are significantly correlated with the community data. Basically, we create distance matrices for the community data and each metadata group and then run Mantel tests for all comparisons. For the community data we calculate Bray-Curtis distances for the community data and Euclidean distances for the metadata. We use the function mantel.test from the ape package and mantel from the vegan package for the analyses.
In summary, we test both mantel.test and mantel on Bray-Curtis distance community distances against Euclidean distances for each metadata group (edaphic, soil_funct, temp_adapt) a) before normalizing and before removing autocorrelated parameters, b) before normalizing and after removing autocorrelated parameters, c) after normalizing and before removing autocorrelated parameters, and d) after normalizing and after removing autocorrelated parameters.
(16S rRNA) Table 4 | Summary of Dissimilarity Correlation Tests using mantel.test from the ape package and mantel from the vegan. P-values in red indicate significance (p-value < 0.05)
Moving on.
Best Subset of Variables
Now we want to know which of the metadata parameters are the most strongly correlated with the community data. For this we use the bioenv function from the vegan package. bioenv—Best Subset of Environmental Variables with Maximum (Rank) Correlation with Community Dissimilarities—finds the best subset of environmental variables, so that the Euclidean distances of scaled environmental variables have the maximum (rank) correlation with community dissimilarities.
Next we will use the metadata set where autocorrelated parameters were removed and the remainder of the parameters were normalized (where applicable based on the Shapiro tests).
We run bioenv against the three groups of metadata parameters. We then run bioenv again, but this time against the individual parameters identified as significantly correlated.
Call:
bioenv(comm = wisconsin(tmp_comm), env = tmp_env, method = "spearman", index = "bray", upto = ncol(tmp_env), metric = "euclidean")
Subset of environmental variables with best correlation to community data.
Correlations: spearman
Dissimilarities: bray
Metric: euclidean
Best model has 1 parameters (max. 15 allowed):
AST
with correlation 0.6808861
Show the results of individual edaphic metadata Mantel tests
$AST
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 1
Significance: 0.001
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.177 0.226 0.271 0.326
Permutation: free
Number of permutations: 999
bioenv found the following edaphic properties significantly correlated with the community data: AST
Call:
bioenv(comm = wisconsin(tmp_comm), env = tmp_env, method = "spearman", index = "bray", upto = ncol(tmp_env), metric = "euclidean")
Subset of environmental variables with best correlation to community data.
Correlations: spearman
Dissimilarities: bray
Metric: euclidean
Best model has 5 parameters (max. 13 allowed):
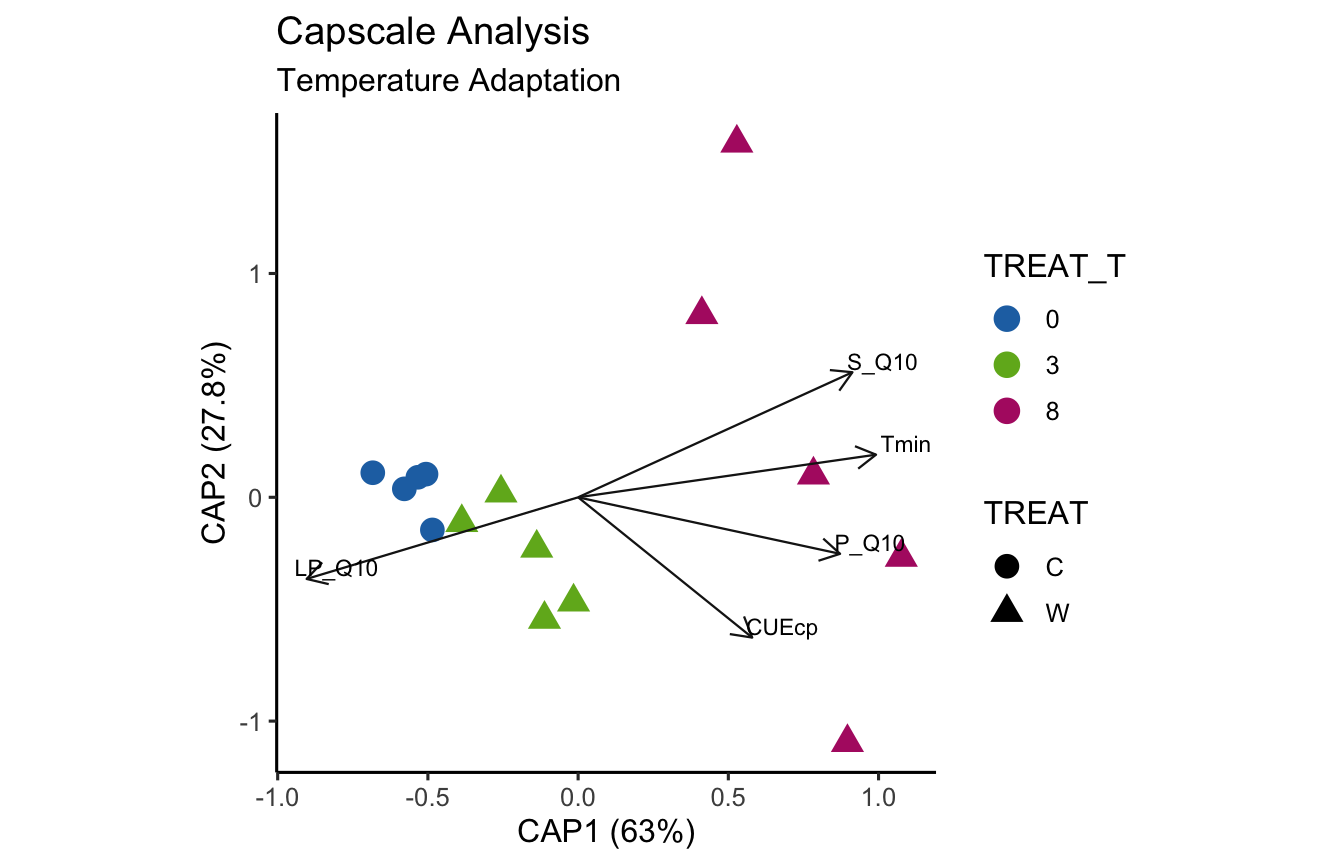
P_Q10 S_Q10 LP_Q10 CUEcp Tmin
with correlation 0.4237093
Show the results of individual temperature adaptation metadata Mantel tests
$CUEcp
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.325
Significance: 0.013
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.188 0.226 0.280 0.331
Permutation: free
Number of permutations: 999
$LP_Q10
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.3772
Significance: 0.005
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.203 0.272 0.306 0.351
Permutation: free
Number of permutations: 999
$P_Q10
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.5177
Significance: 0.001
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.158 0.211 0.261 0.309
Permutation: free
Number of permutations: 999
$S_Q10
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.4395
Significance: 0.001
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.159 0.205 0.252 0.301
Permutation: free
Number of permutations: 999
$Tmin
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.4044
Significance: 0.005
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.207 0.267 0.305 0.359
Permutation: free
Number of permutations: 999
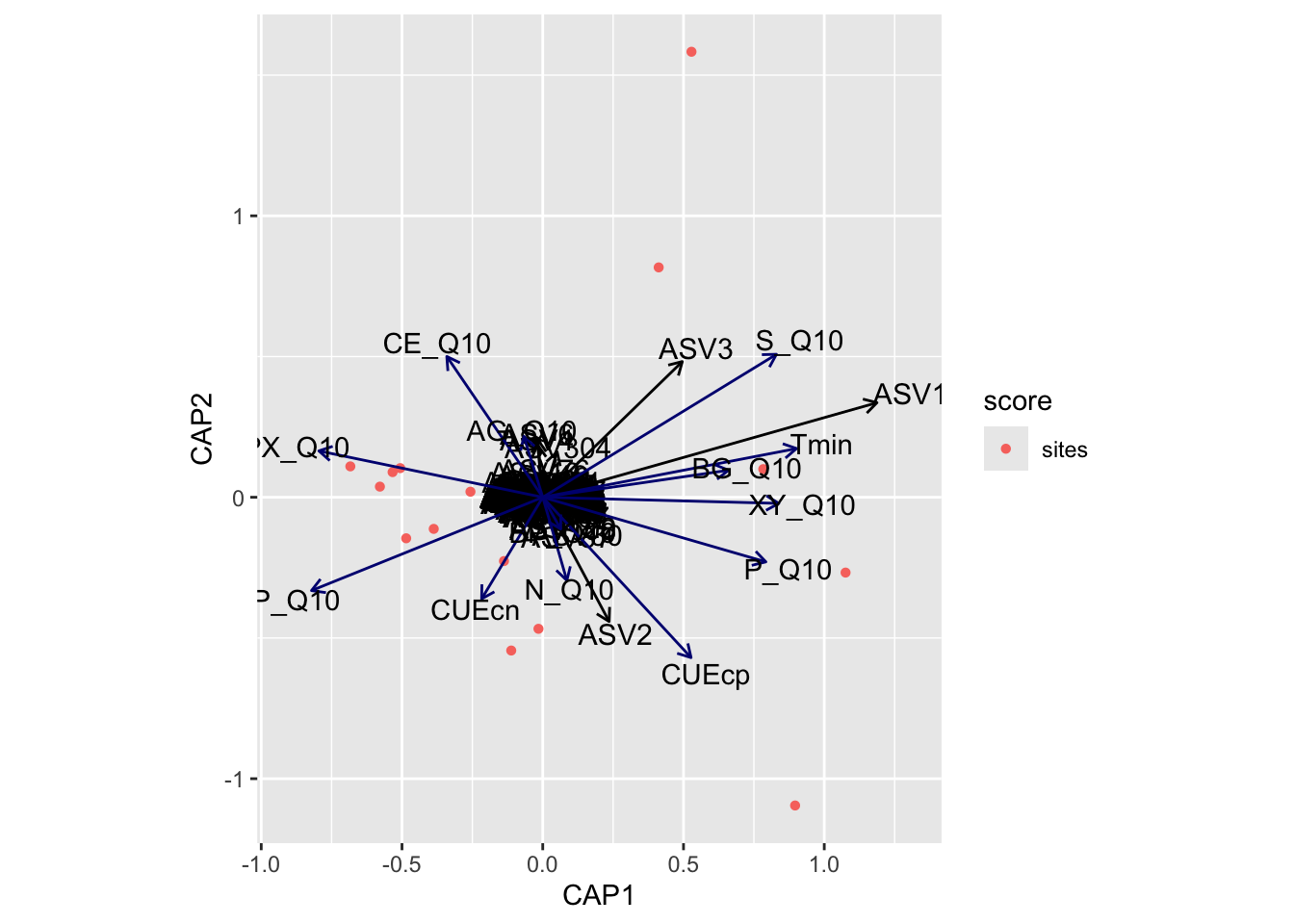
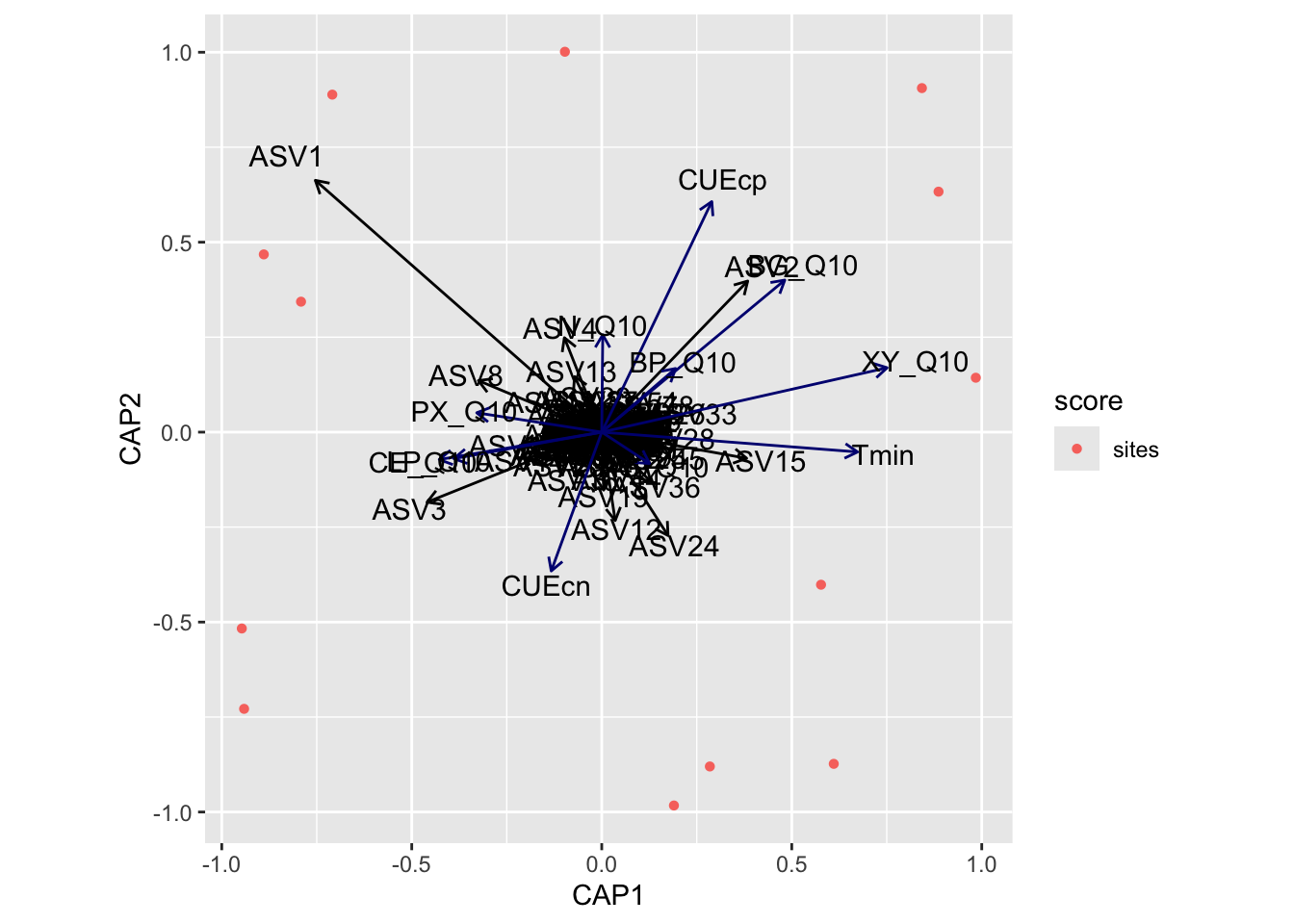
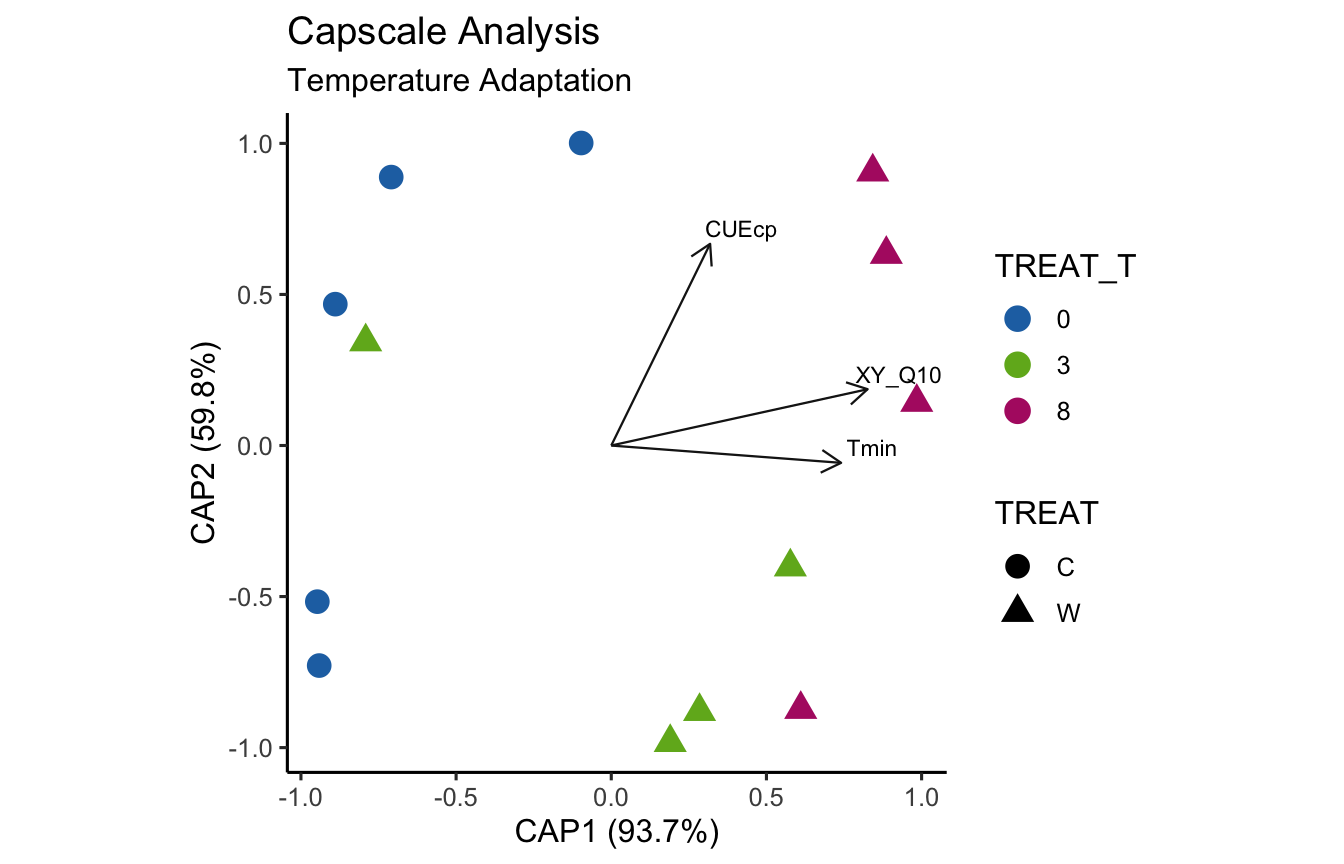
bioenv found the following temperature adaptations significantly correlated with the community data: CUEcp, LP_Q10, P_Q10, S_Q10, Tmin
Distance-based Redundancy
Now we turn our attention to distance-based redundancy analysis (dbRDA), an ordination method similar to Redundancy Analysis (rda) but it allows non-Euclidean dissimilarity indices, such as Manhattan or Bray–Curtis distance. For this, we use capscale from the vegan package. capscale is a constrained versions of metric scaling (principal coordinates analysis), which are based on the Euclidean distance but can be used, and are more useful, with other dissimilarity measures. The functions can also perform unconstrained principal coordinates analysis, optionally using extended dissimilarities.
For each of the three metadata subsets, we perform the following steps:
Run rankindex to compare metadata and community dissimilarity indices for gradient detection. This will help us select the best dissimilarity metric to use.
Run capscale for distance-based redundancy analysis.
Run envfit to fit environmental parameters onto the ordination. This function basically calculates correlation scores between the metadata parameters and the ordination axes.
Select metadata parameters significant for bioenv (see above) and/or envfit analyses.
euc man gow bra kul
0.3171263 0.4336927 0.1028302 0.4336927 0.4336927
Let’s run capscale using Bray-Curtis. Note, we have 15 metadata parameters in this group but, for some reason, capscale only works with 13 parameters. This may have to do with degrees of freedom?
Call: capscale(formula = tmp_comm ~ AST + H2O + N + P + Fe + K + ECEC + pH
+ NH4 + NO3 + PO4 + DOC + DOCN, data = tmp_md, distance = "bray")
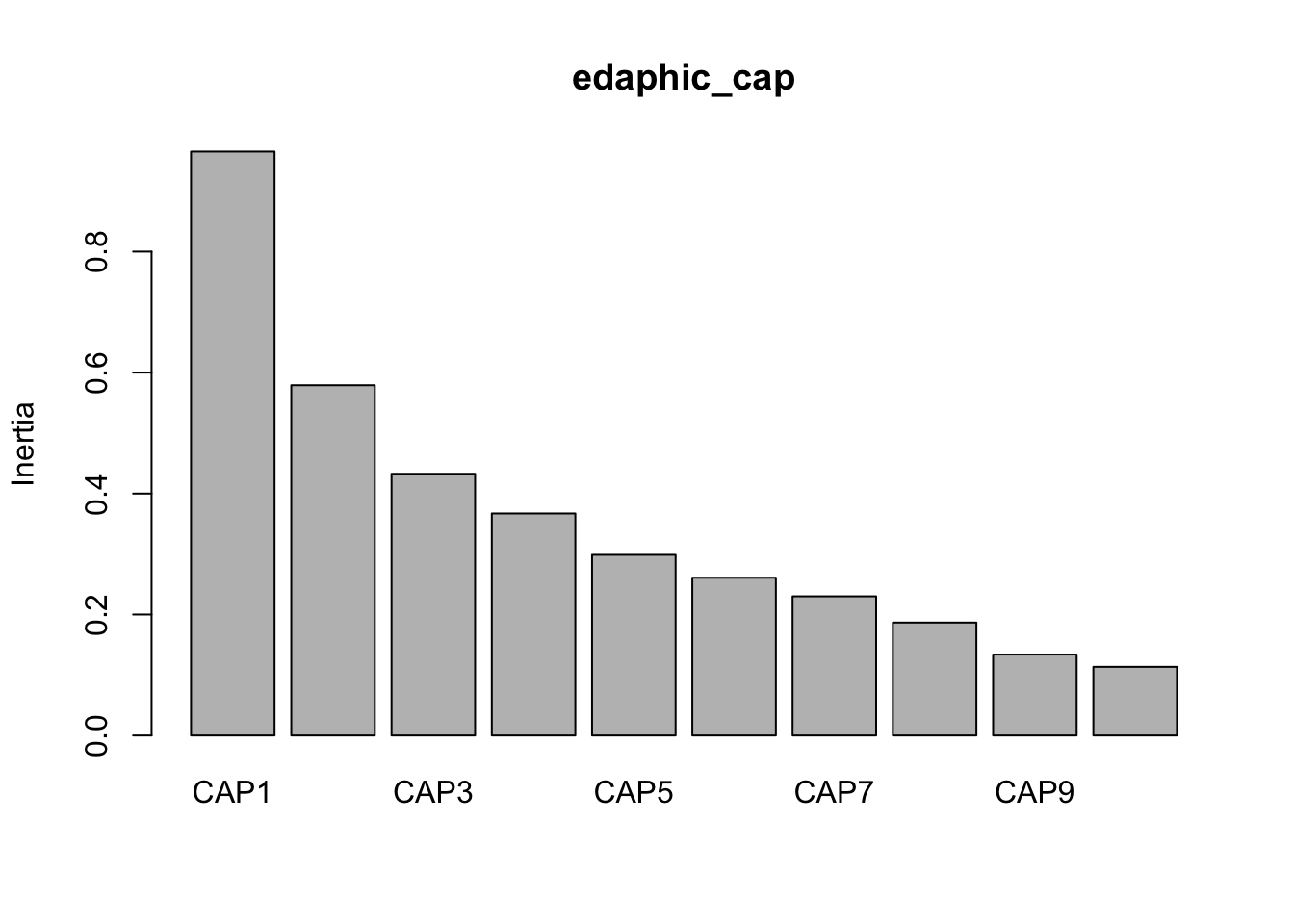
Inertia Proportion Rank
Total 1.67890 1.00000
Constrained 1.63037 0.97110 13
Unconstrained 0.04853 0.02890 1
Inertia is squared Bray distance
-- NOTE:
Species scores projected from 'tmp_comm'
Eigenvalues for constrained axes:
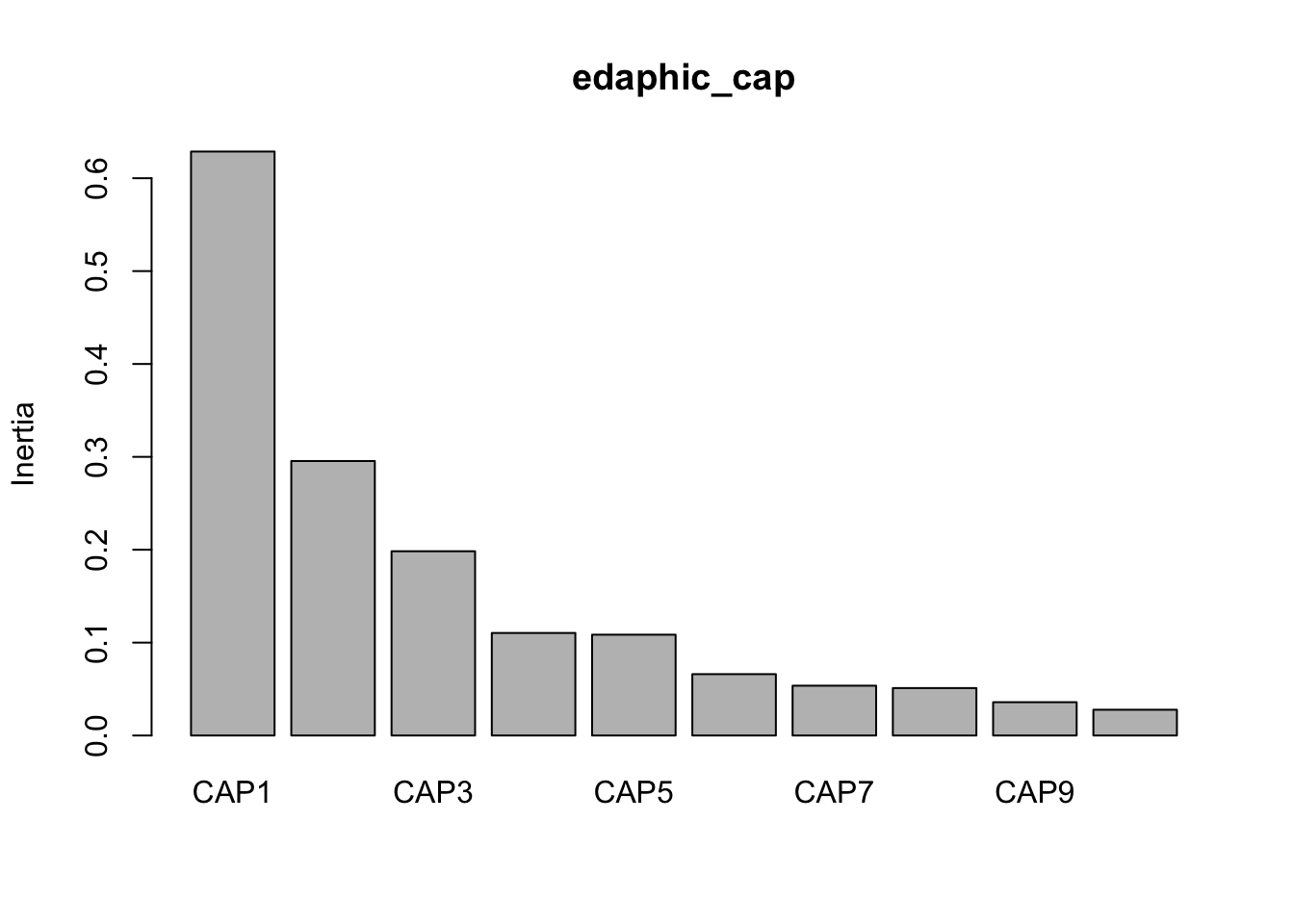
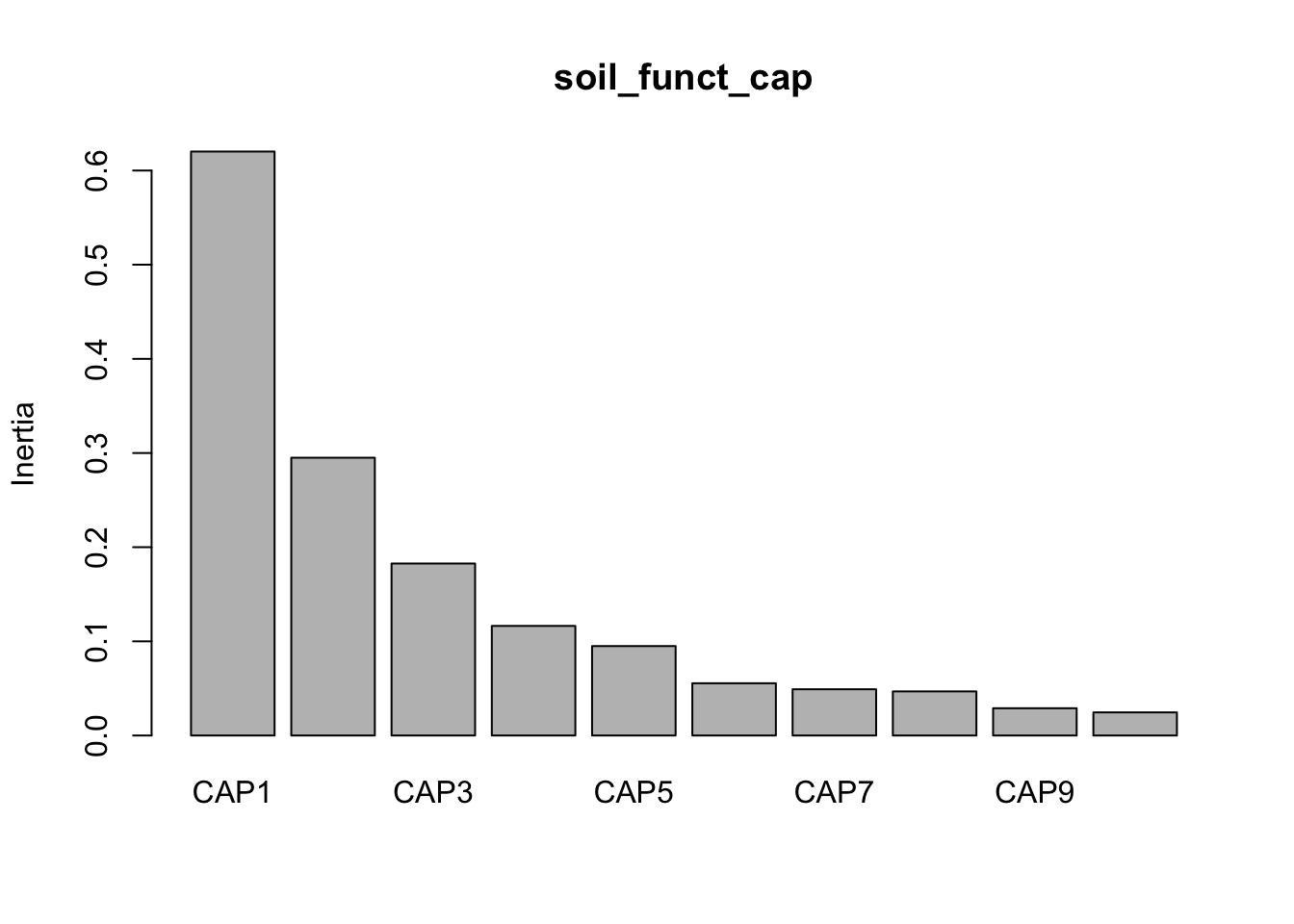
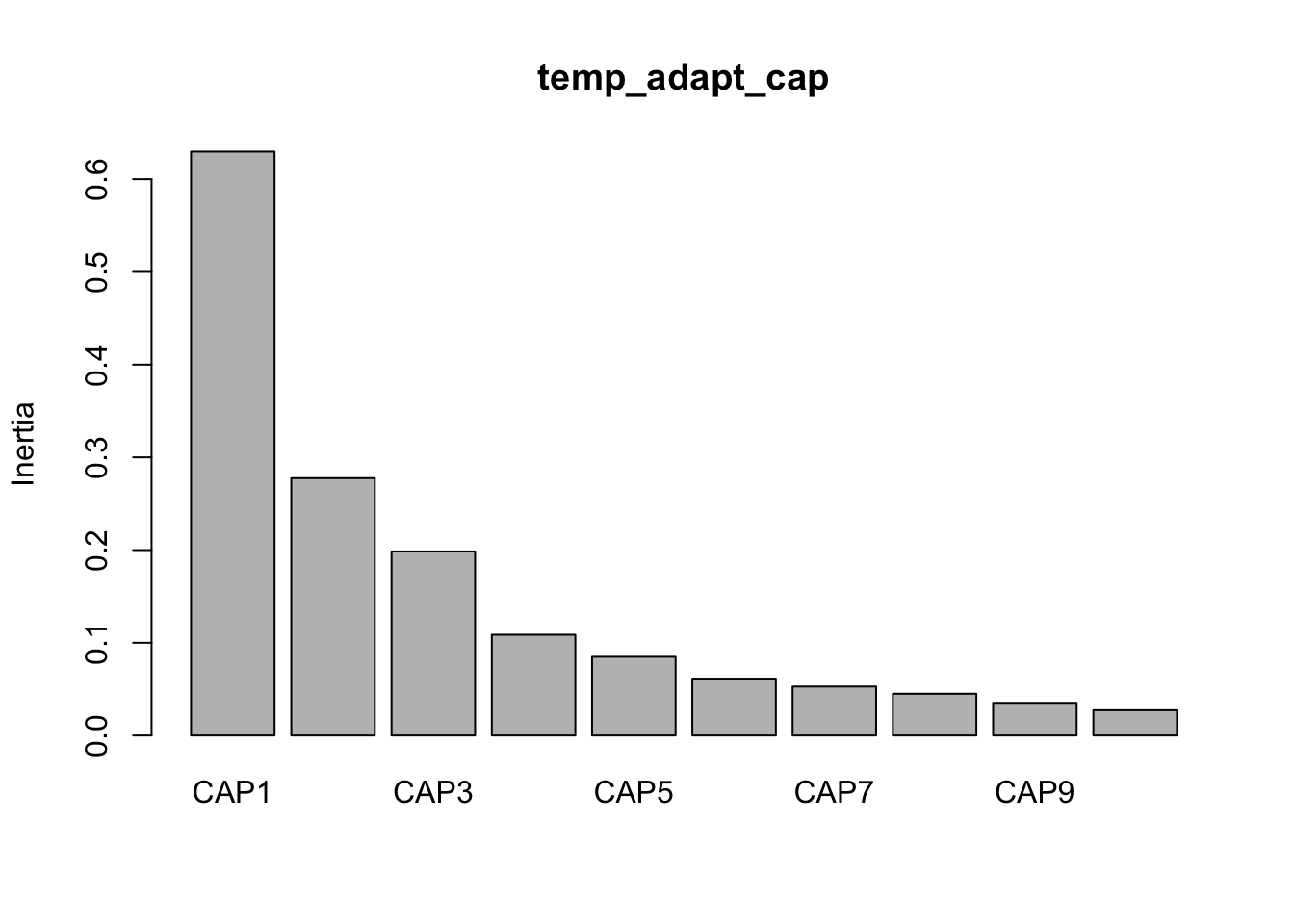
CAP1 CAP2 CAP3 CAP4 CAP5 CAP6 CAP7 CAP8 CAP9 CAP10 CAP11
0.6287 0.2955 0.1984 0.1104 0.1085 0.0661 0.0536 0.0511 0.0359 0.0277 0.0247
CAP12 CAP13
0.0218 0.0080
Eigenvalues for unconstrained axes:
MDS1
0.04853
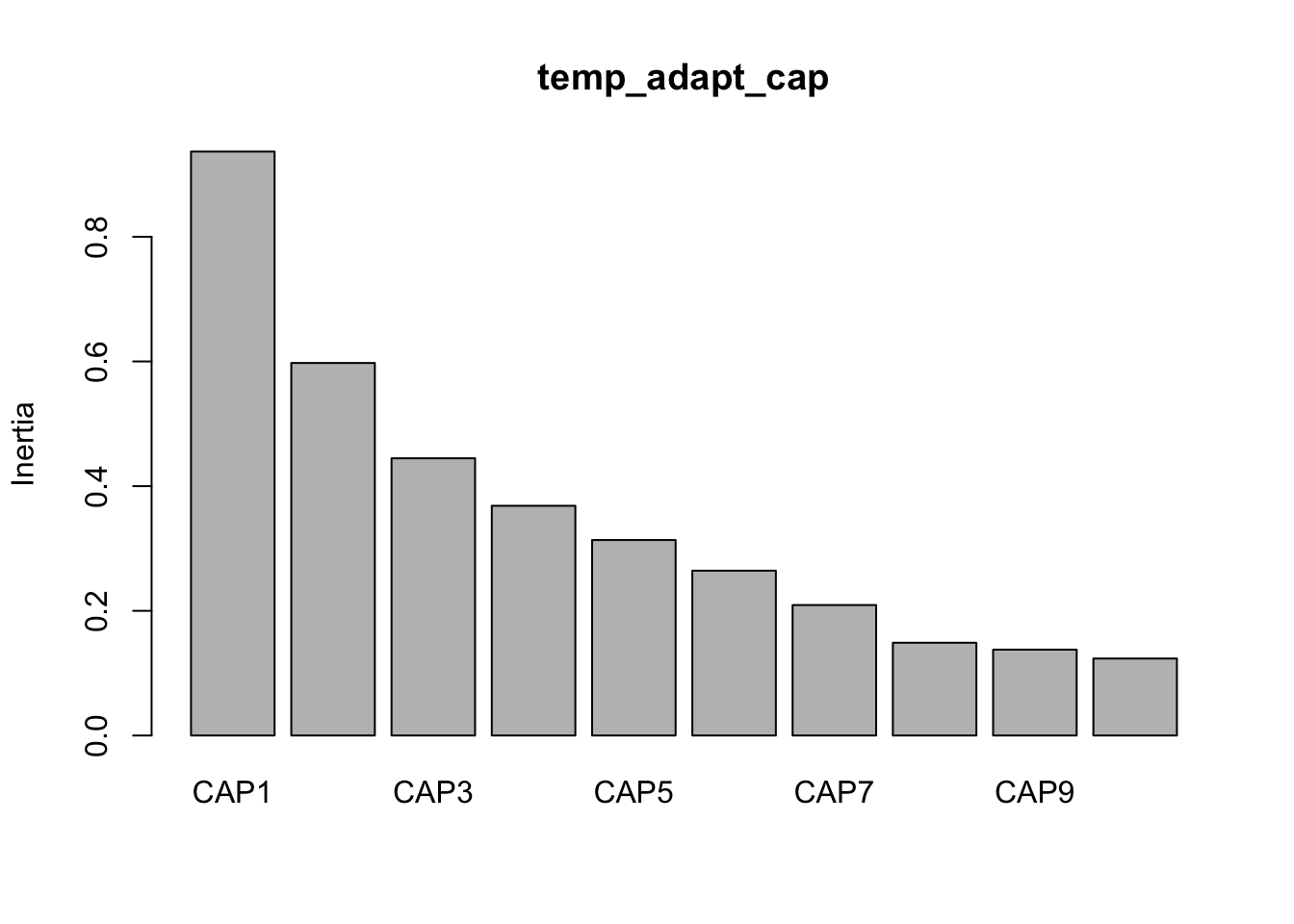
Now we can look at the variance against each principal component.
And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The ggplot function autoplot stores these data in a more accessible way than the raw results from capscale
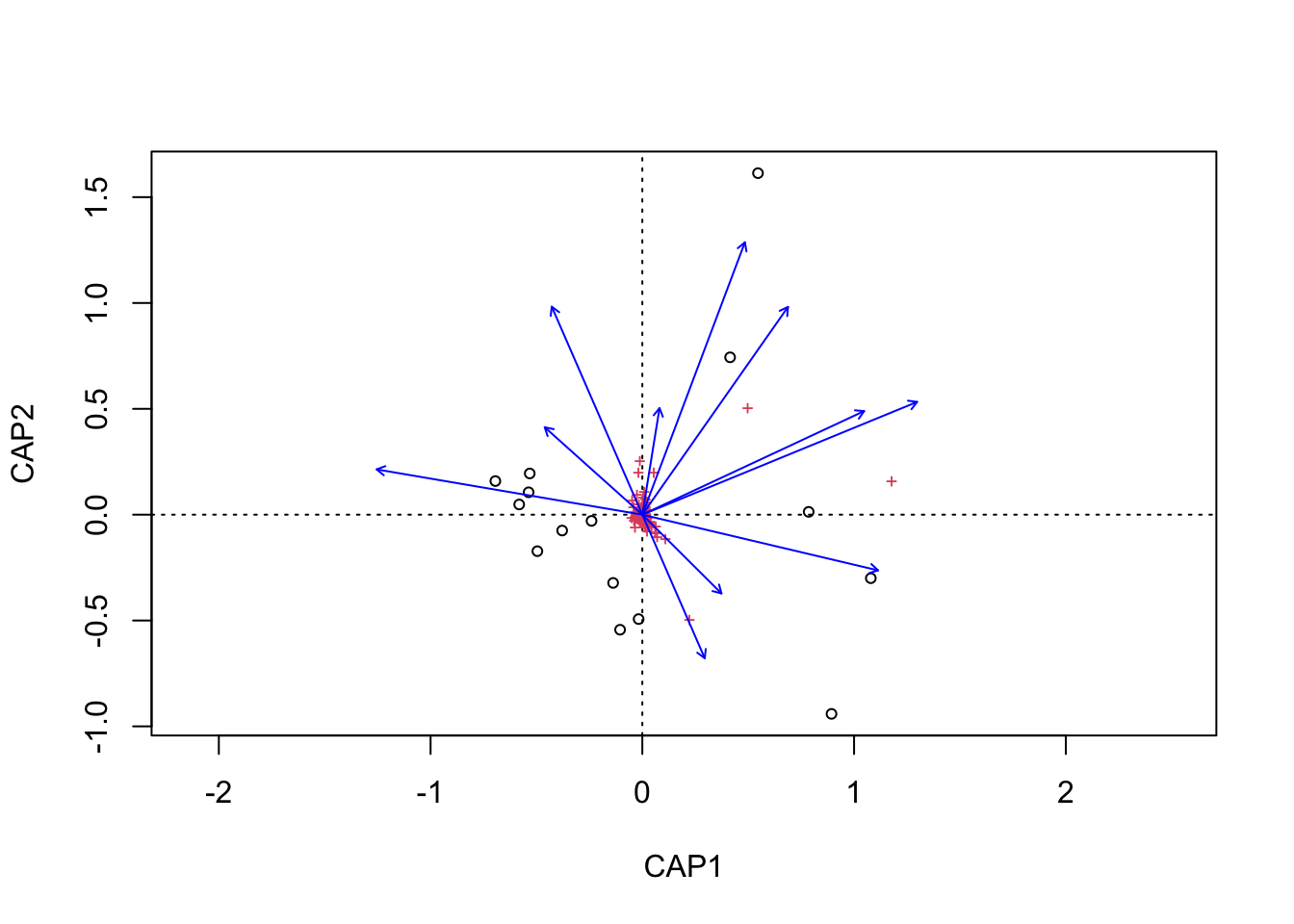
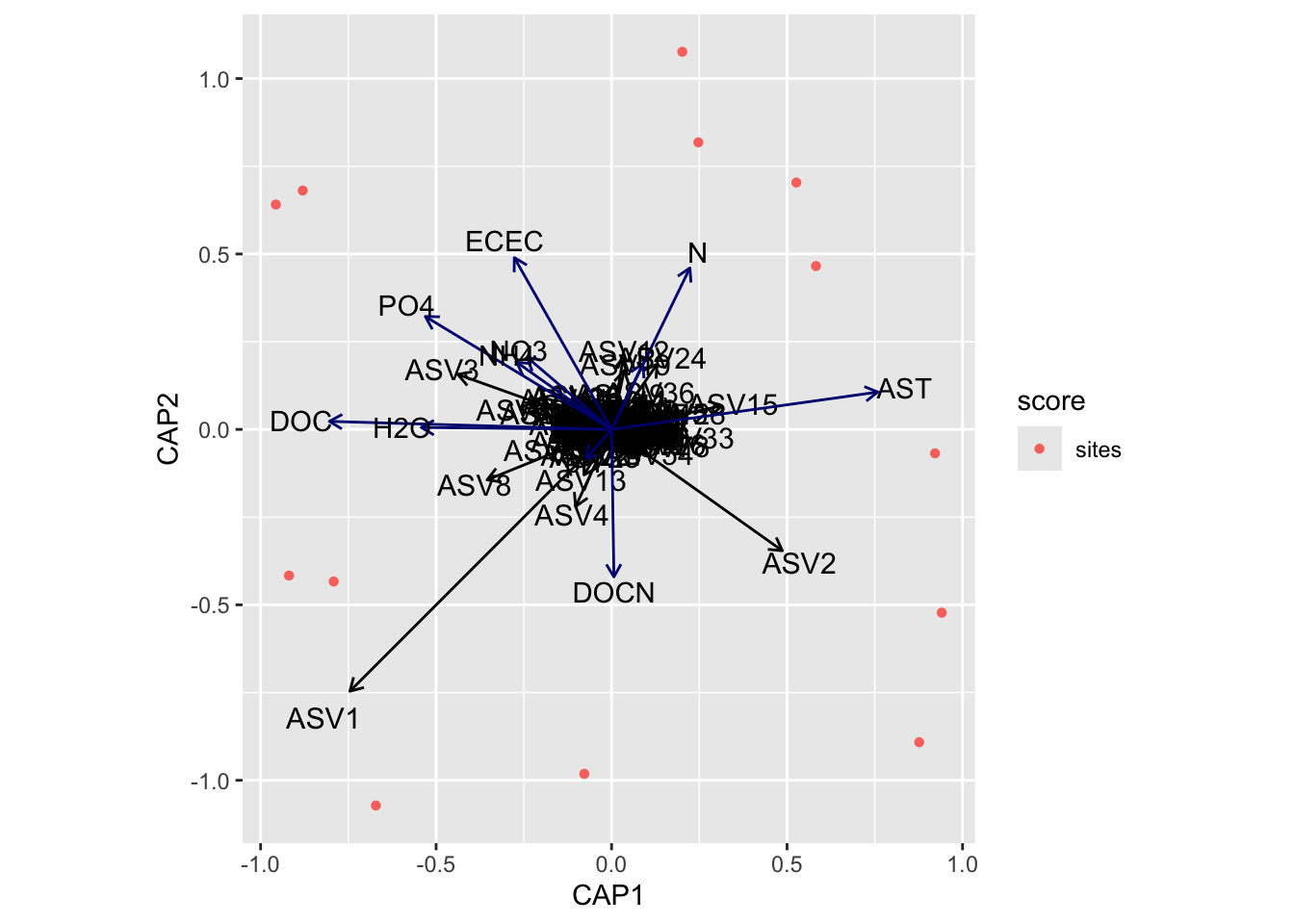
Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function scores which gets species or site scores from the ordination.
Show the code
tmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")edaphic_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")
Now we have a new data frame that contains sample details and capscale values.
SampleID PLOT TREAT TREAT_T PAIR CAP1 CAP2
1 P10_D00_010_C0E P10 C 0 E -0.51023381 0.16071056348
2 P02_D00_010_C0A P02 C 0 A -0.48448722 -0.19055366181
3 P04_D00_010_C0B P04 C 0 B -0.58218619 0.02325887477
4 P06_D00_010_C0C P06 C 0 C -0.53370906 0.09973976767
5 P08_D00_010_C0D P08 C 0 D -0.68492943 0.13143857676
6 P01_D00_010_W3A P01 W 3 A -0.13286443 -0.28661650806
7 P03_D00_010_W3B P03 W 3 B -0.25415783 0.00002258673
8 P05_D00_010_W3C P05 W 3 C -0.38829574 -0.08695154559
9 P07_D00_010_W3D P07 W 3 D -0.01360582 -0.49632449304
10 P09_D00_010_W3E P09 W 3 E -0.10639798 -0.54219706978
11 P01_D00_010_W8A P01 W 8 A 0.78689725 0.02214444812
12 P03_D00_010_W8B P03 W 8 B 0.89574754 -0.92786190328
13 P05_D00_010_W8C P05 W 8 C 1.07945322 -0.28874425650
14 P07_D00_010_W8D P07 W 8 D 0.41234456 0.75597843926
15 P09_D00_010_W8E P09 W 8 E 0.51642495 1.62595618128
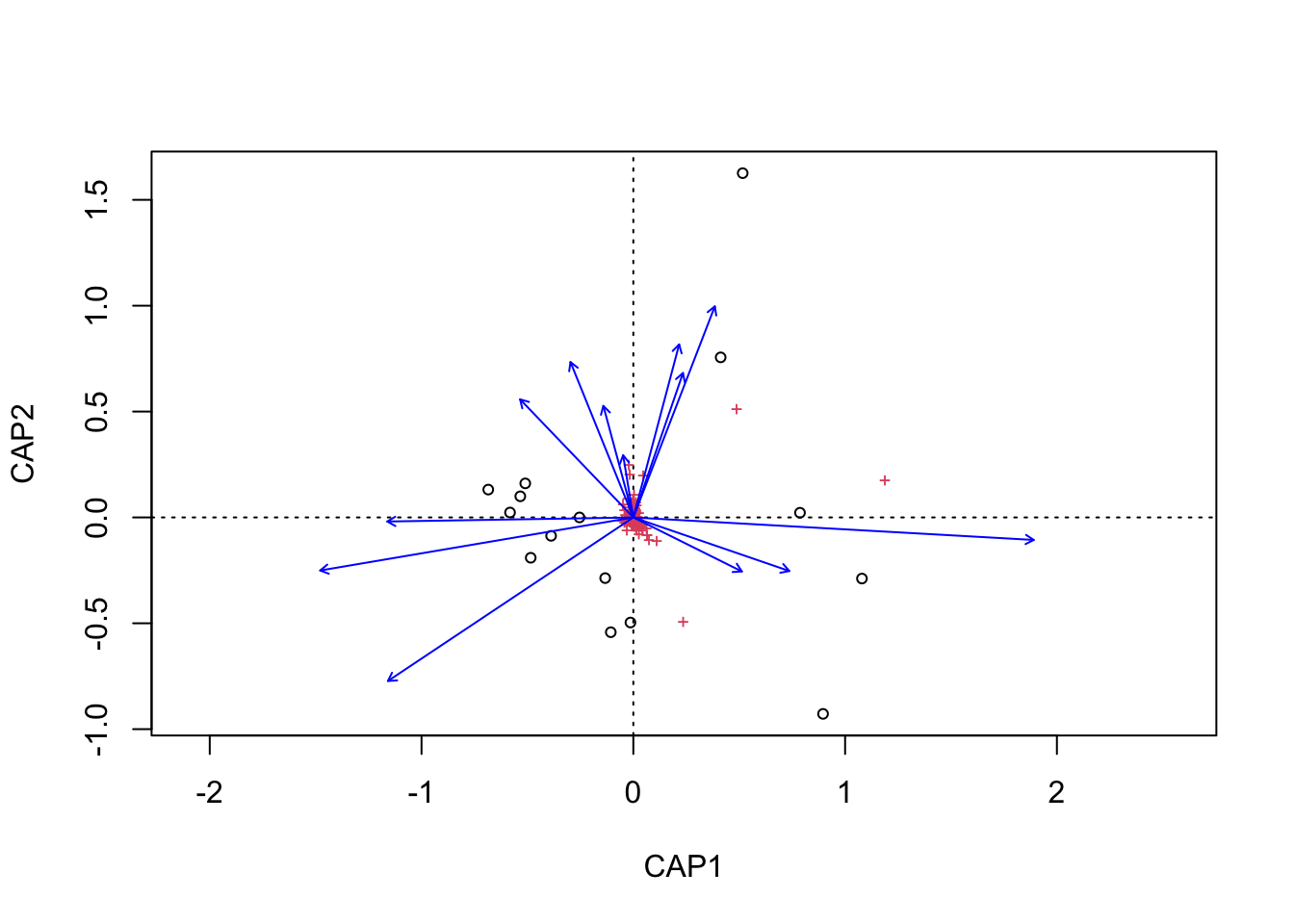
We can then do the same with the metadata vectors. Here though we only need the scores and parameter name.
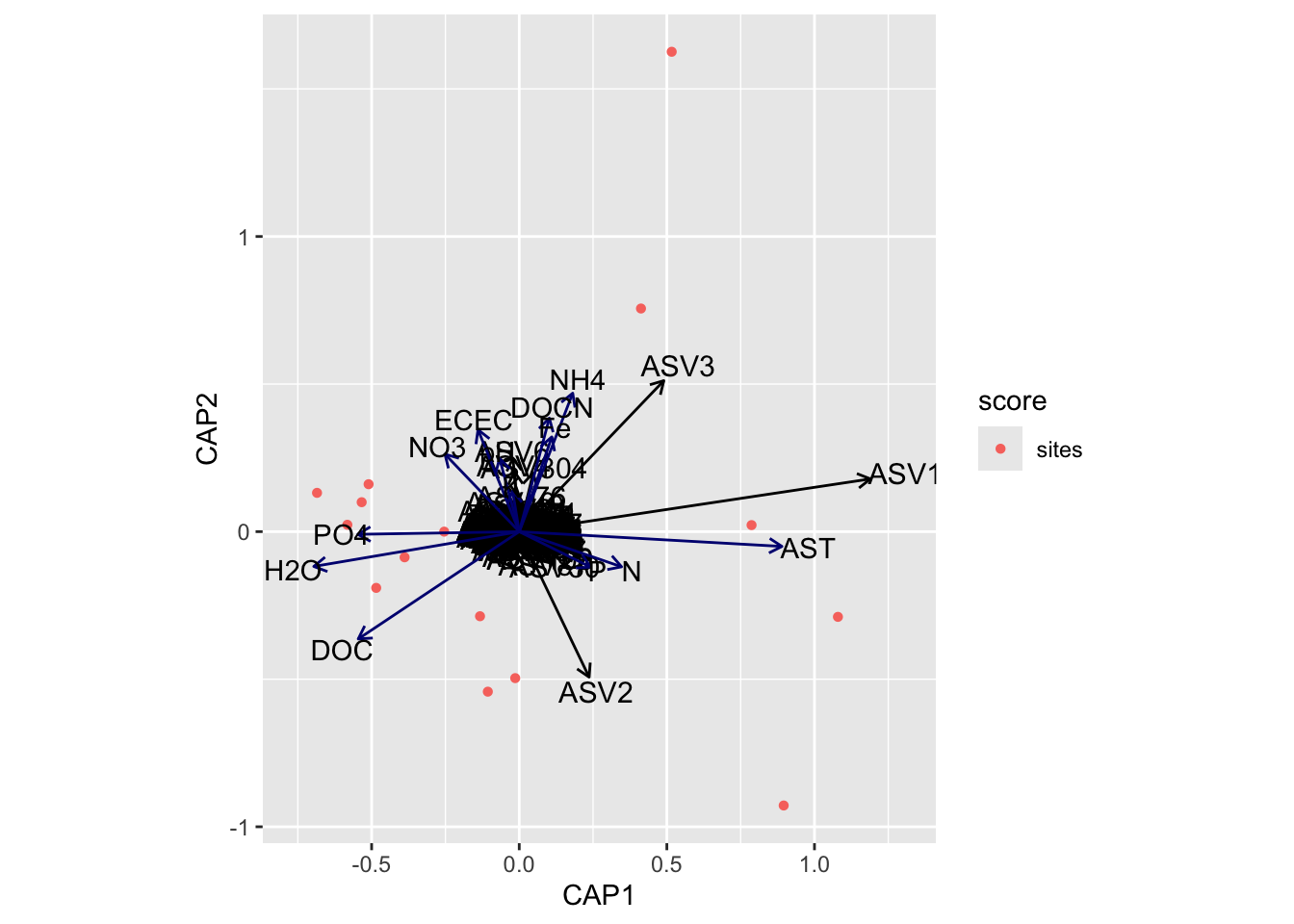
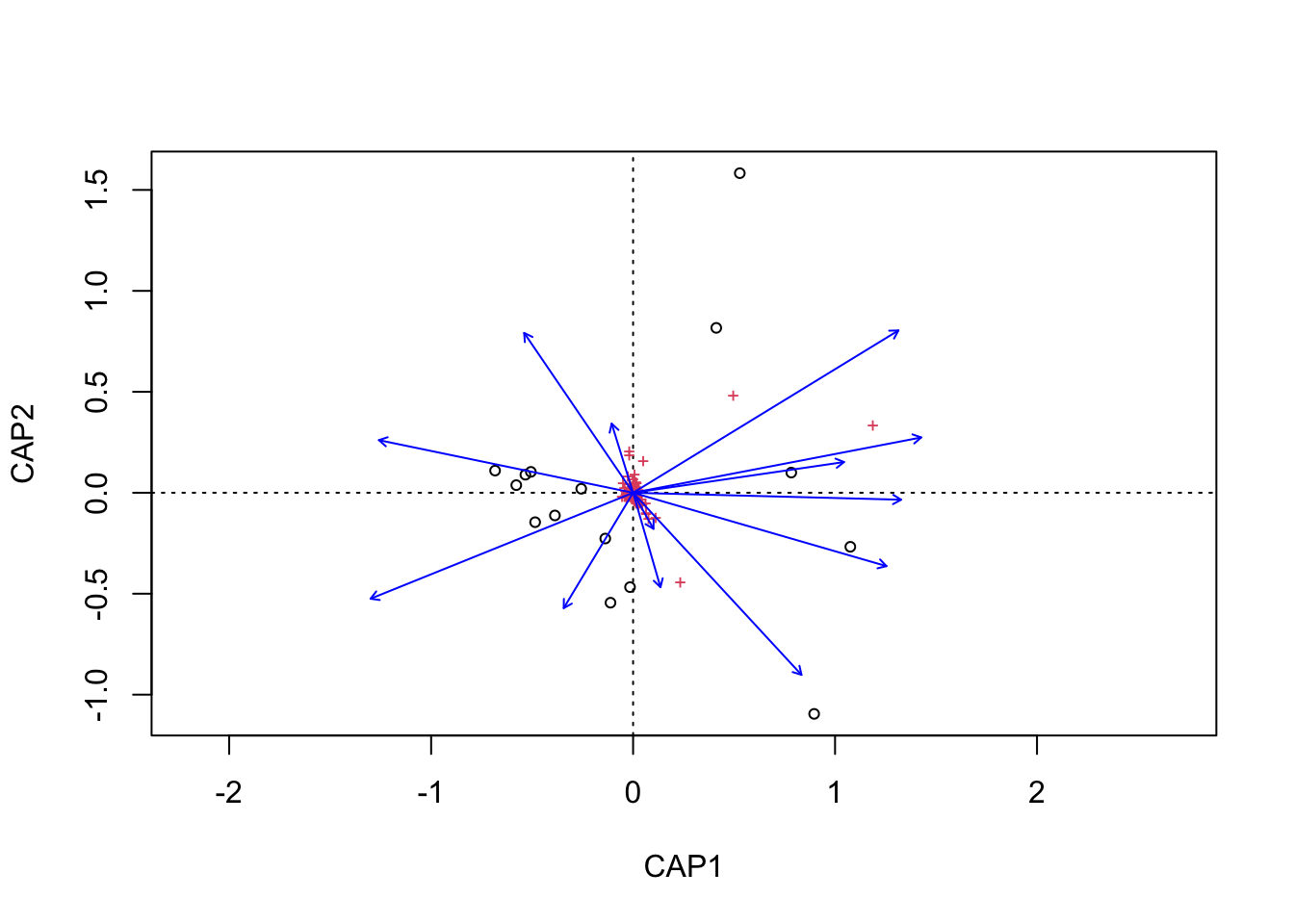
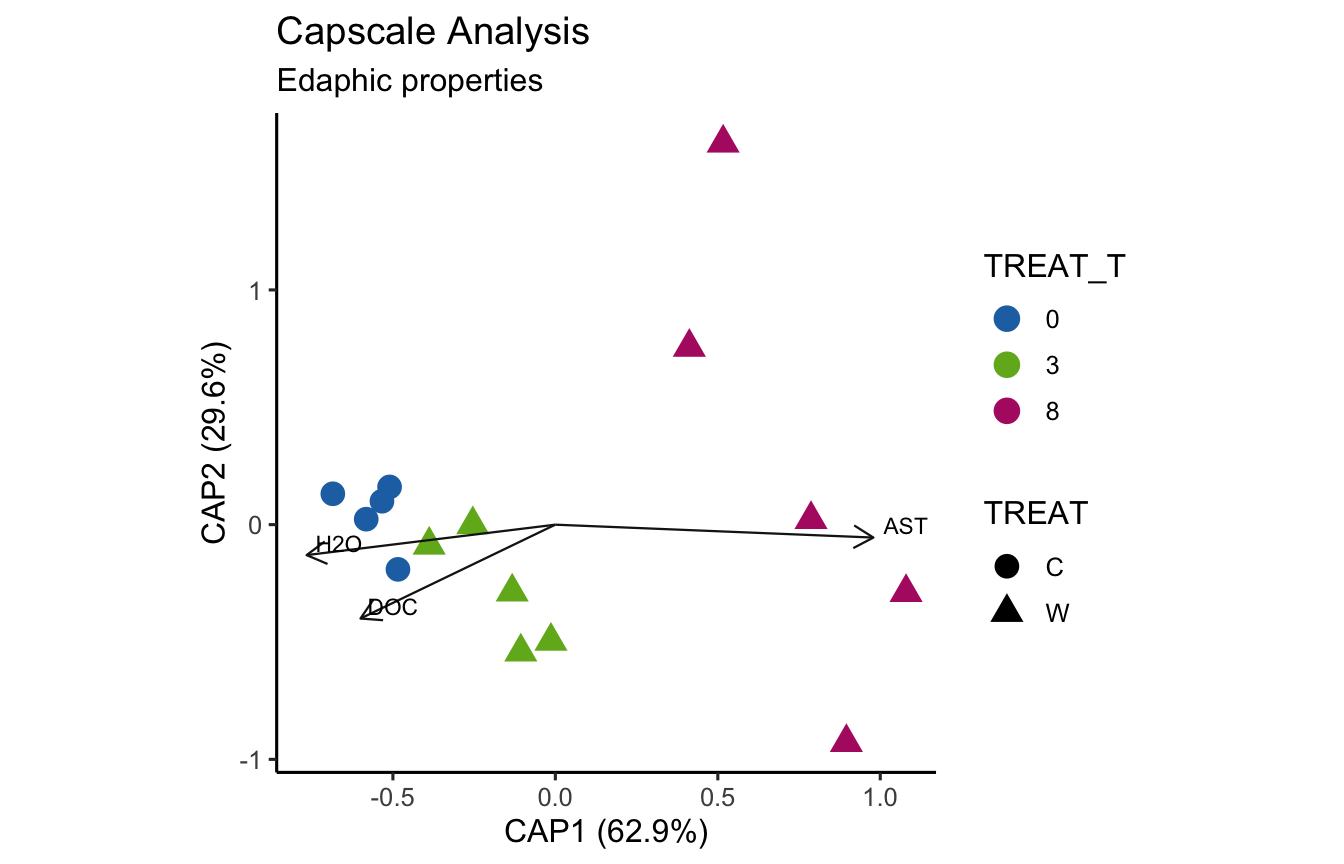
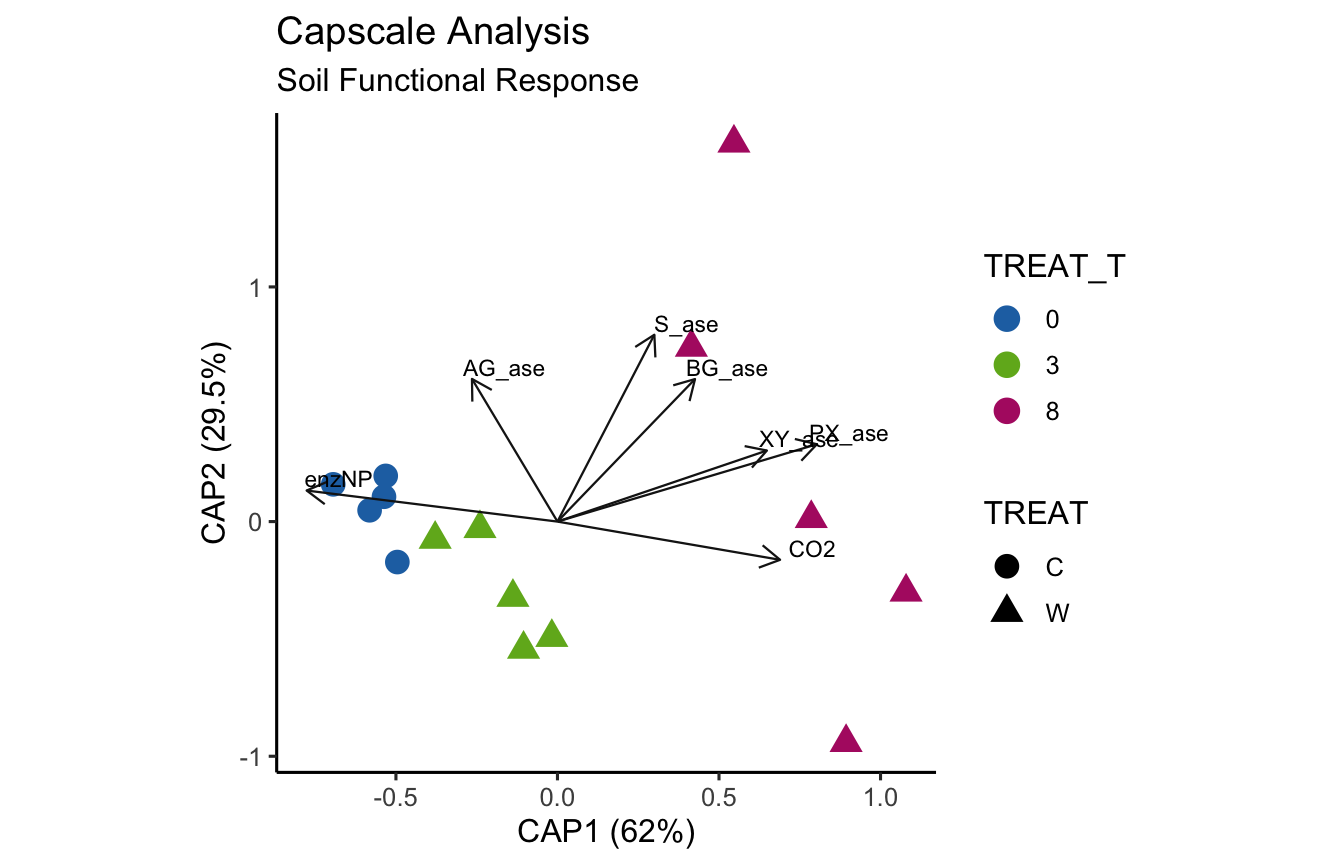
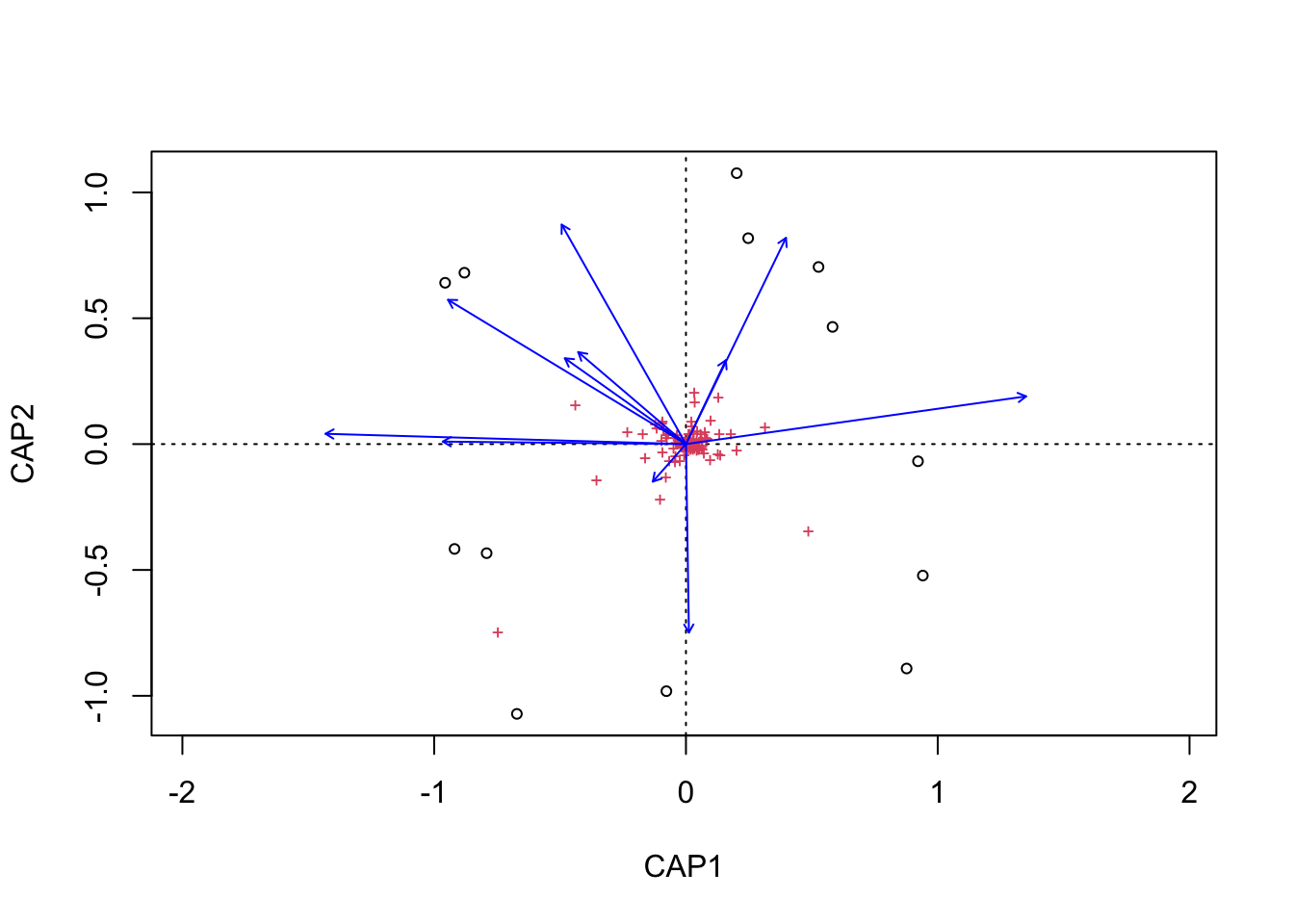
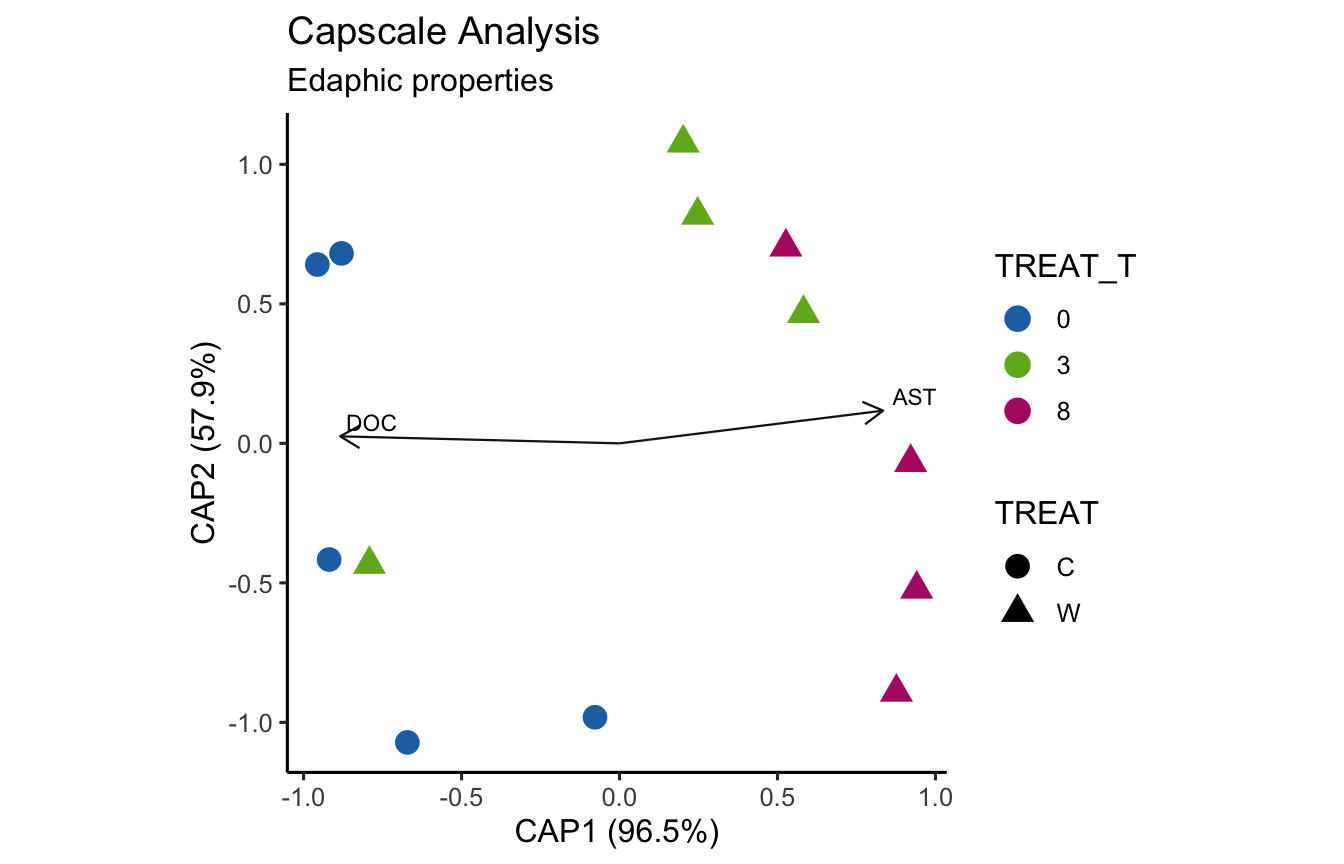
envfit found that AST, H2O, DOC were significantly correlated.
Now let’s see if the same parameters are significant for the envfit and bioenv analyses.
[1] "Significant parameters from bioenv analysis."
[1] "AST"
_____________________________________
[1] "Significant parameters from envfit analysis."
[1] "AST" "H2O" "DOC"
_____________________________________
[1] "Found in bioenv but not envfit."
character(0)
_____________________________________
[1] "Found in envfit but not bioenv."
[1] "H2O" "DOC"
_____________________________________
[1] "Found in envfit and bioenv."
[1] "AST" "H2O" "DOC"
euc man gow bra kul
0.5840452 0.7341800 0.2168775 0.7341800 0.7341800
Let’s run capscale using Bray-Curtis. Note, we have 11 metadata parameters in this group but, for some reason, capscale only works with 13 parameters. This may have to do with degrees of freedom?
Now we can look at the variance against each principal component.
And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The ggplot function autoplot stores these data in a more accessible way than the raw results from capscale
Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function scores which gets species or site scores from the ordination.
Show the code
tmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")soil_funct_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")
Now we have a new data frame that contains sample details and capscale values.
SampleID PLOT TREAT TREAT_T PAIR CAP1 CAP2
1 P10_D00_010_C0E P10 C 0 E -0.53163678 0.19467717
2 P02_D00_010_C0A P02 C 0 A -0.49565928 -0.17246578
3 P04_D00_010_C0B P04 C 0 B -0.58149488 0.04815608
4 P06_D00_010_C0C P06 C 0 C -0.53700662 0.10581563
5 P08_D00_010_C0D P08 C 0 D -0.69408551 0.15871504
6 P01_D00_010_W3A P01 W 3 A -0.13789308 -0.32241637
7 P03_D00_010_W3B P03 W 3 B -0.23991258 -0.02936566
8 P05_D00_010_W3C P05 W 3 C -0.37861217 -0.07503400
9 P07_D00_010_W3D P07 W 3 D -0.01748558 -0.49326034
10 P09_D00_010_W3E P09 W 3 E -0.10498973 -0.54355896
11 P01_D00_010_W8A P01 W 8 A 0.78557974 0.01332924
12 P03_D00_010_W8B P03 W 8 B 0.89342218 -0.94105534
13 P05_D00_010_W8C P05 W 8 C 1.07904998 -0.30014637
14 P07_D00_010_W8D P07 W 8 D 0.41449841 0.74331915
15 P09_D00_010_W8E P09 W 8 E 0.54622591 1.61329051
We can then do the same with the metadata vectors. Here though we only need the scores and parameter name.
euc man gow bra kul
0.2732117 0.3330085 0.3456355 0.3330085 0.3330085
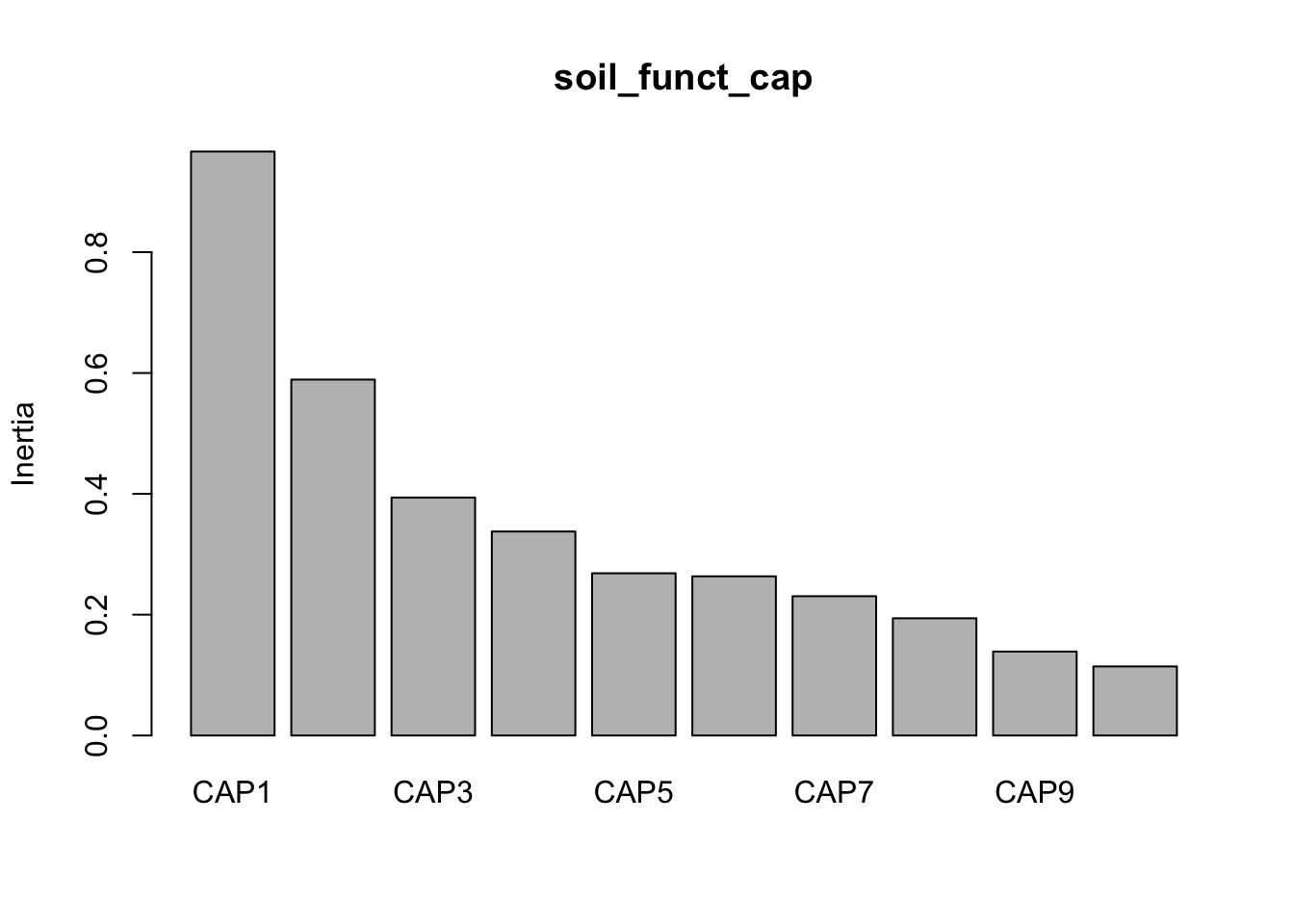
Let’s run capscale using Bray-Curtis. Note, we have 13 metadata parameters in this group but, for some reason, capscale only works with 13 parameters. This may have to do with degrees of freedom?
Now we can look at the variance against each principal component.
And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The ggplot function autoplot stores these data in a more accessible way than the raw results from capscale
Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function scores which gets species or site scores from the ordination.
Show the code
tmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")temp_adapt_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")
Now we have a new data frame that contains sample details and capscale values.
SampleID PLOT TREAT TREAT_T PAIR CAP1 CAP2
1 P10_D00_010_C0E P10 C 0 E -0.50625375 0.10348626
2 P02_D00_010_C0A P02 C 0 A -0.48509579 -0.14580527
3 P04_D00_010_C0B P04 C 0 B -0.57845671 0.03776429
4 P06_D00_010_C0C P06 C 0 C -0.53336078 0.08936059
5 P08_D00_010_C0D P08 C 0 D -0.68345909 0.10974979
6 P01_D00_010_W3A P01 W 3 A -0.13776351 -0.22674265
7 P03_D00_010_W3B P03 W 3 B -0.25679729 0.01972197
8 P05_D00_010_W3C P05 W 3 C -0.38722889 -0.11236725
9 P07_D00_010_W3D P07 W 3 D -0.01518359 -0.46741081
10 P09_D00_010_W3E P09 W 3 E -0.11194937 -0.54460136
11 P01_D00_010_W8A P01 W 8 A 0.78329851 0.09997647
12 P03_D00_010_W8B P03 W 8 B 0.89642936 -1.09522700
13 P05_D00_010_W8C P05 W 8 C 1.07566246 -0.26764258
14 P07_D00_010_W8D P07 W 8 D 0.41185806 0.81673870
15 P09_D00_010_W8E P09 W 8 E 0.52830039 1.58299886
We can then do the same with the metadata vectors. Here though we only need the scores and parameter name.
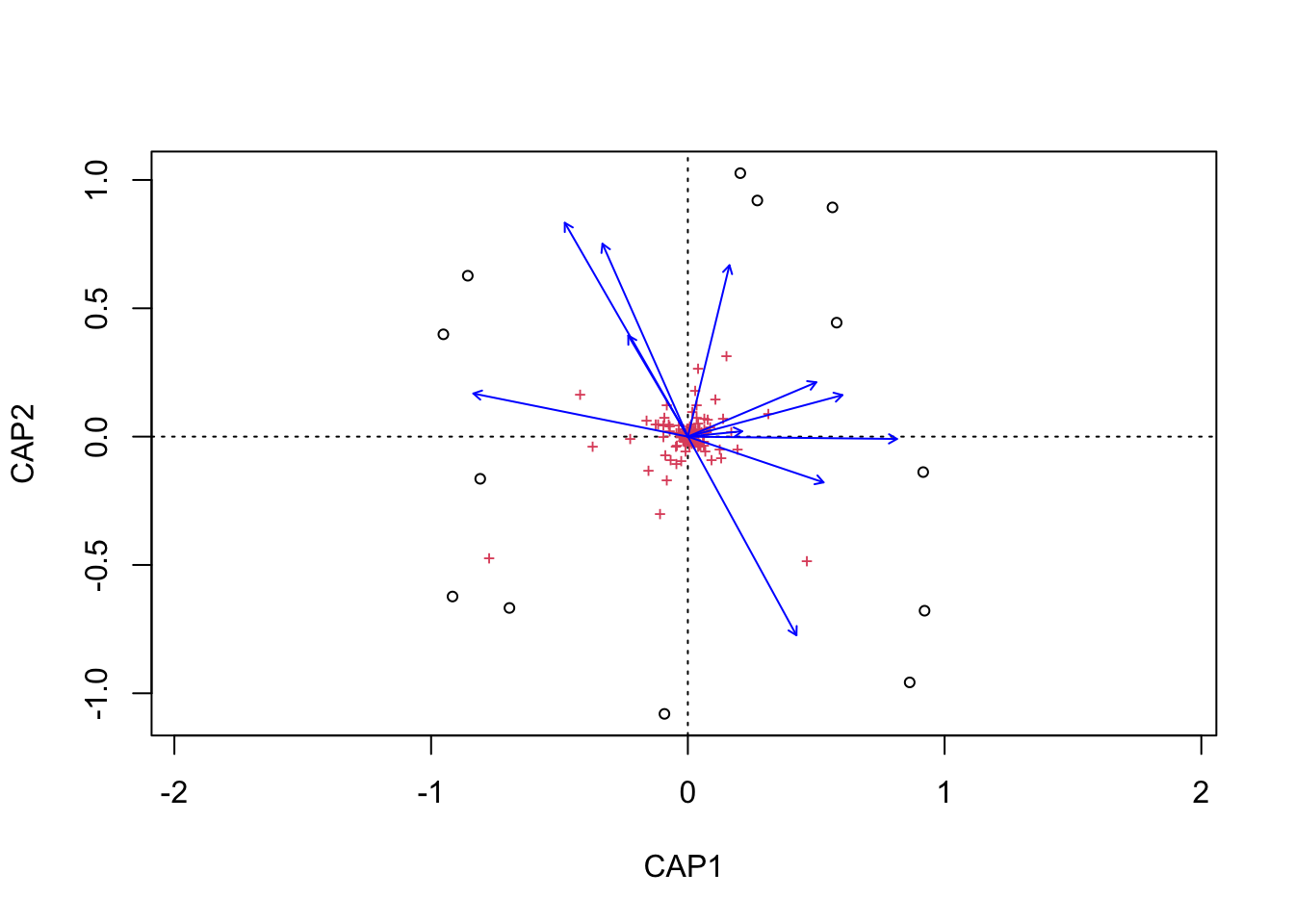
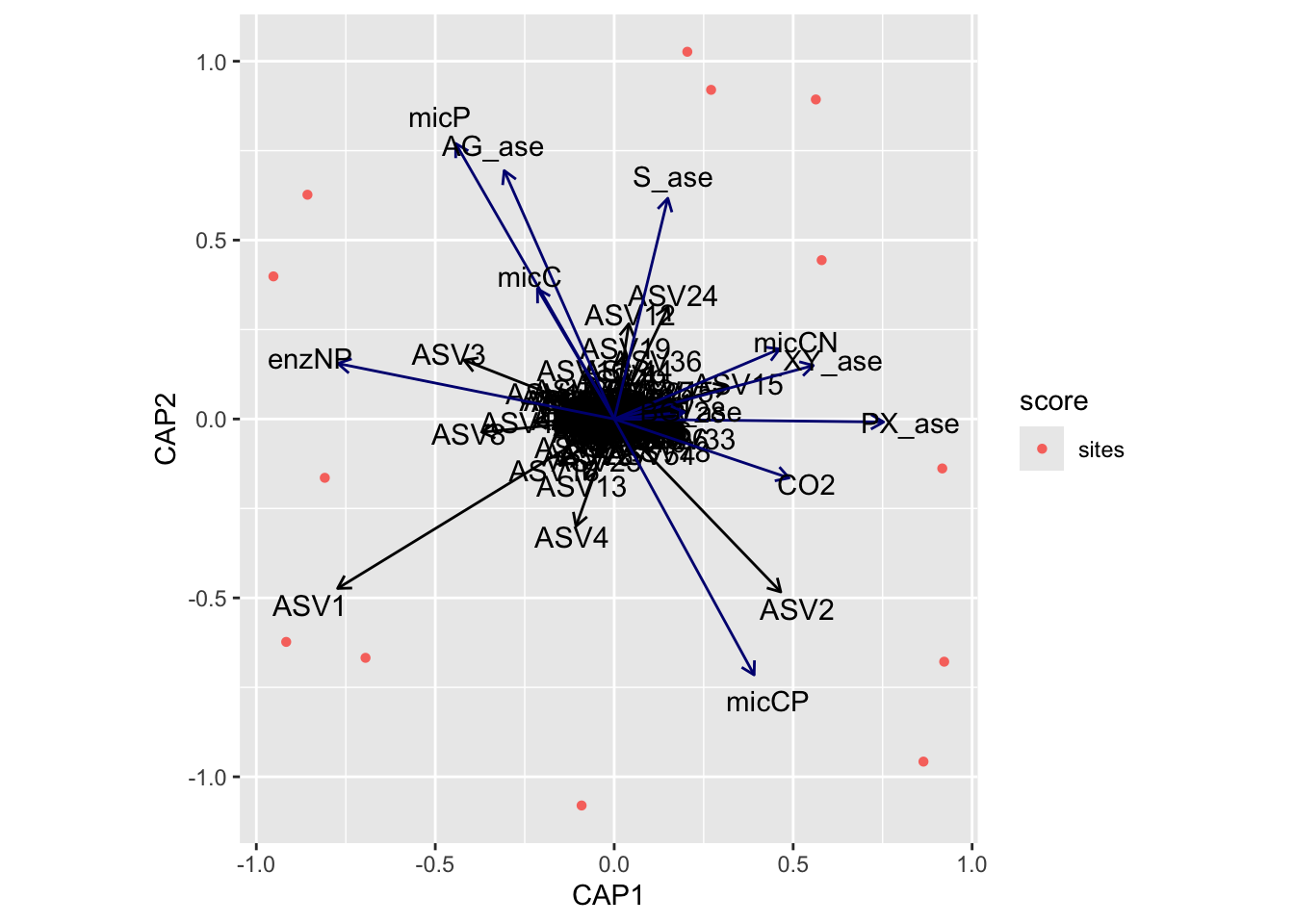
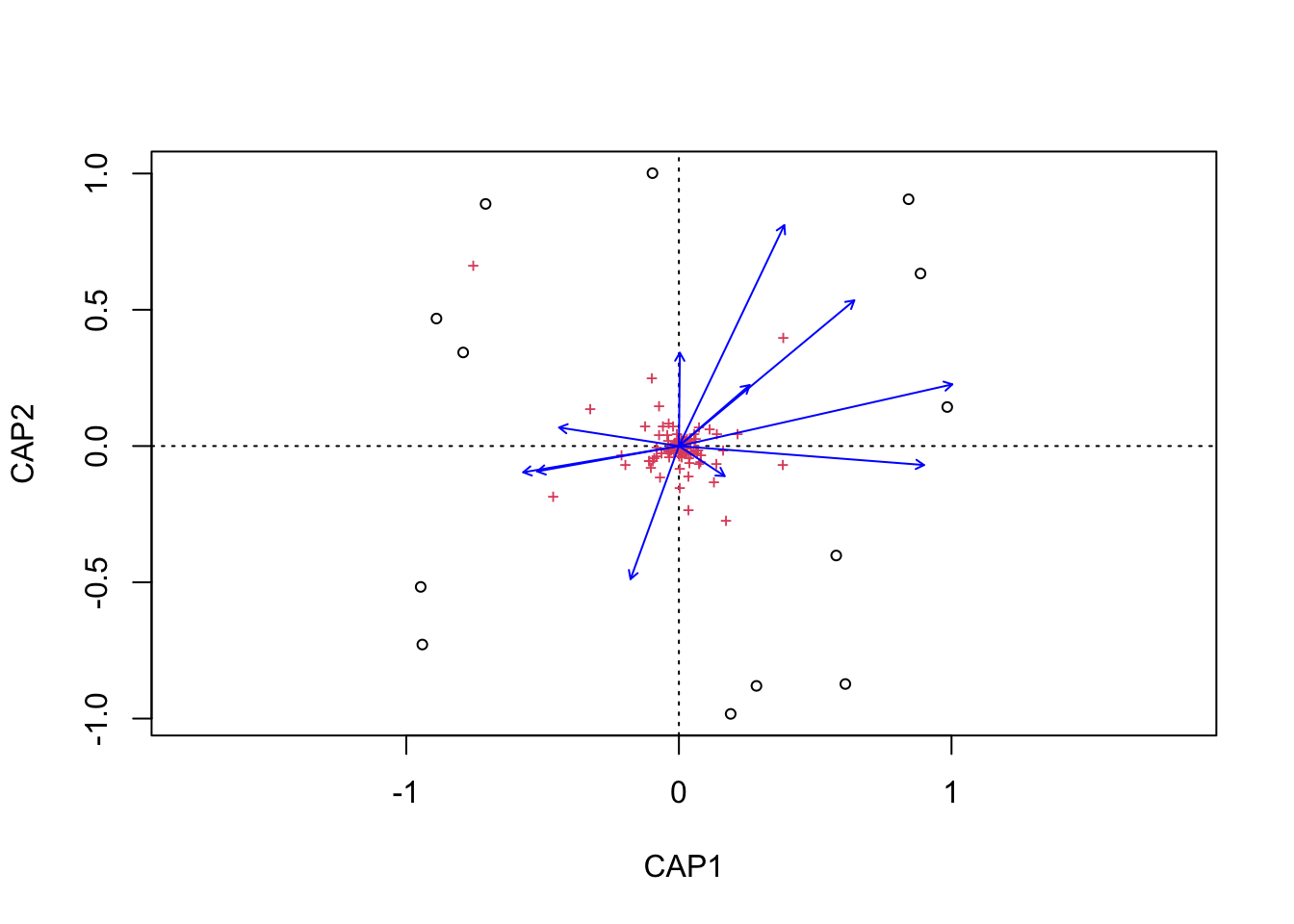
(16S rRNA) Figure 7 | Distance-based Redundancy Analysis (db-RDA) of PIME filtered data based on Bray-Curtis dissimilarity showing the relationships between community composition change versus edaphic properties.
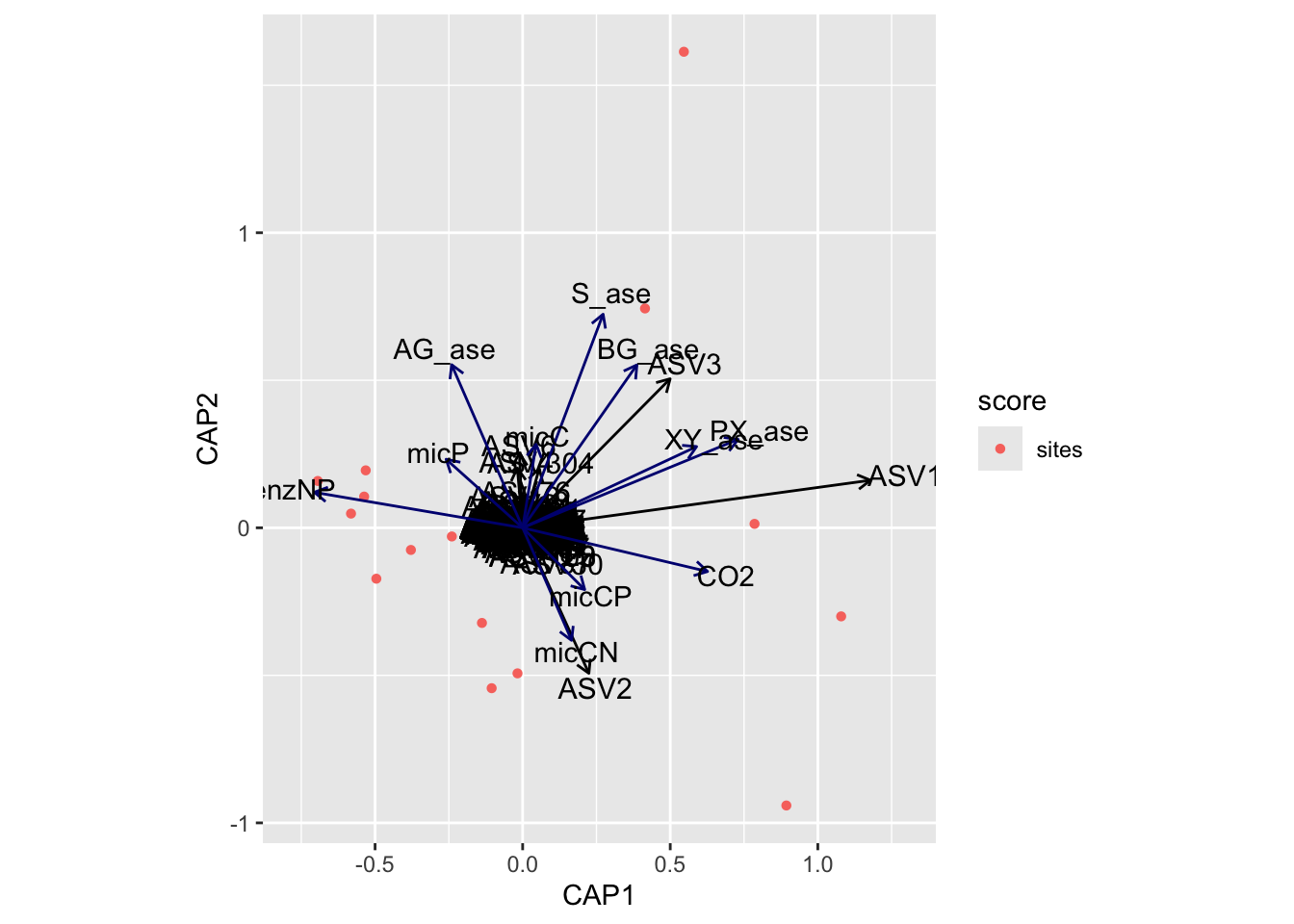
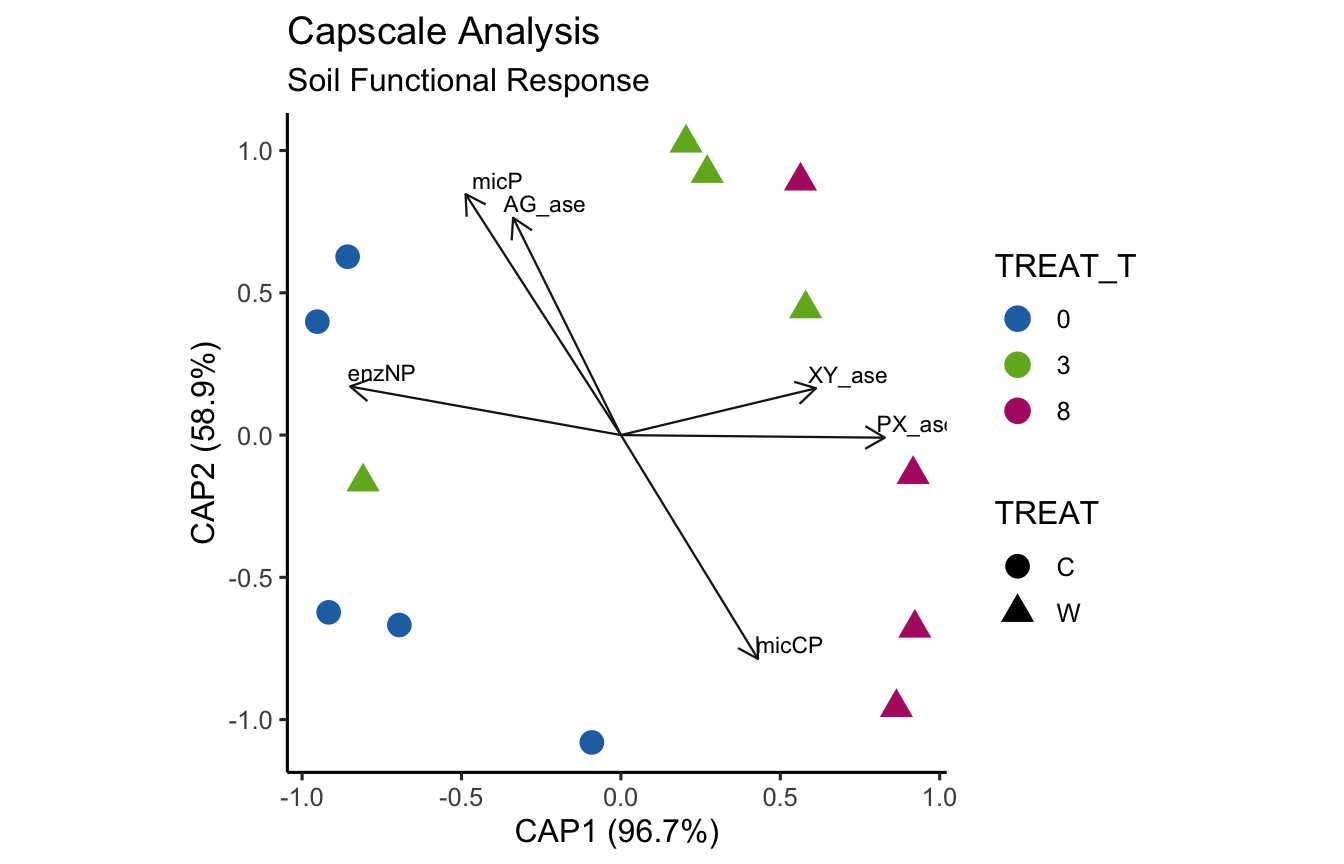
(16S rRNA) Figure 8 | Distance-based Redundancy Analysis (db-RDA) of PIME filtered data based on Bray-Curtis dissimilarity showing the relationships between community composition change versus microbial functional response.
(16S rRNA) Figure 9 | Distance-based Redundancy Analysis (db-RDA) of PIME filtered data based on Bray-Curtis dissimilarity showing the relationships between community composition change versus temperature adaptation.
ITS
Data Formating
Transform ps Objects
The first step is to transform the phyloseq objects.
Before proceeding, we need to test each parameter in the metadata to see which ones are and are not normally distributed. For that, we use the Shapiro-Wilk Normality Test. Here we only need one of the metadata files.
Show the results of each normality test for metadata parameters
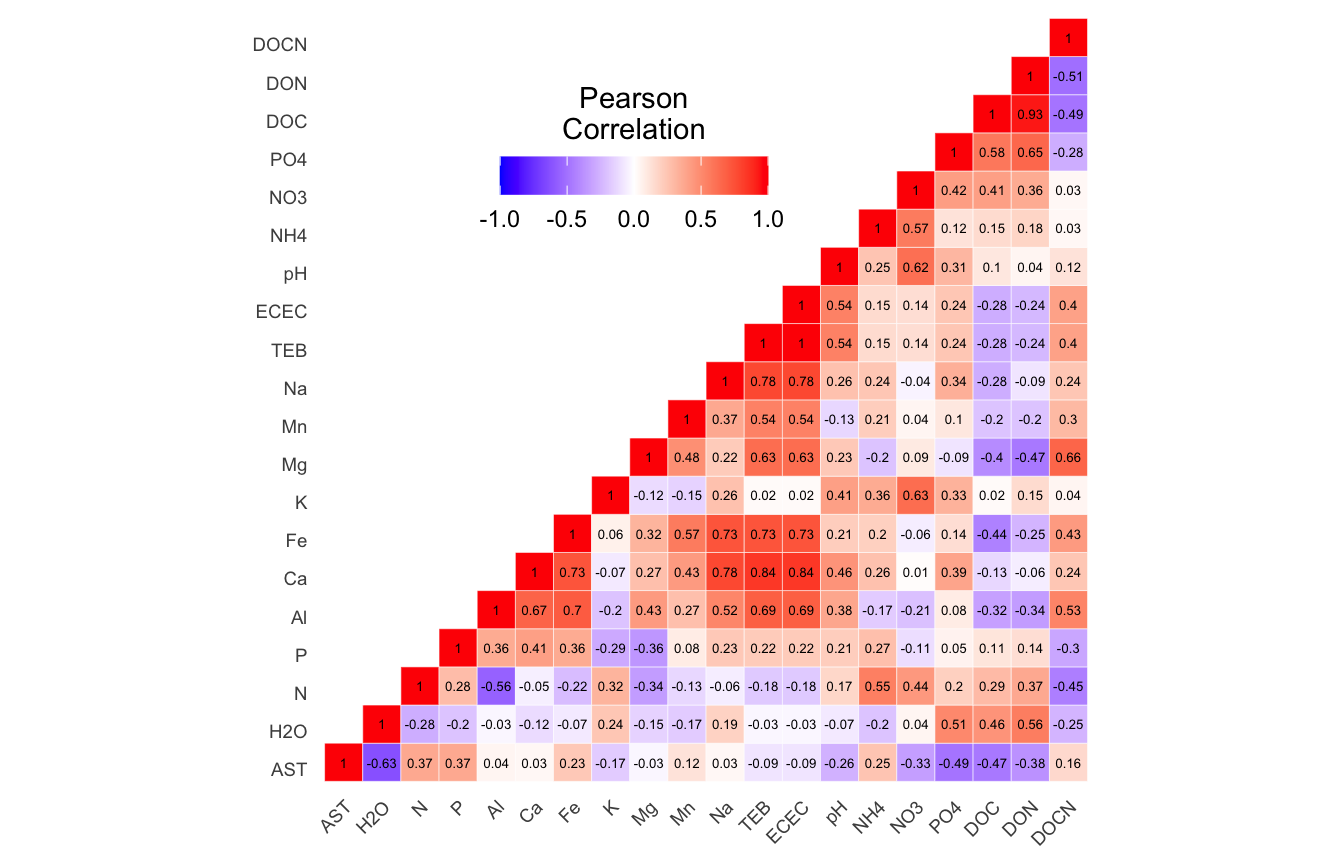
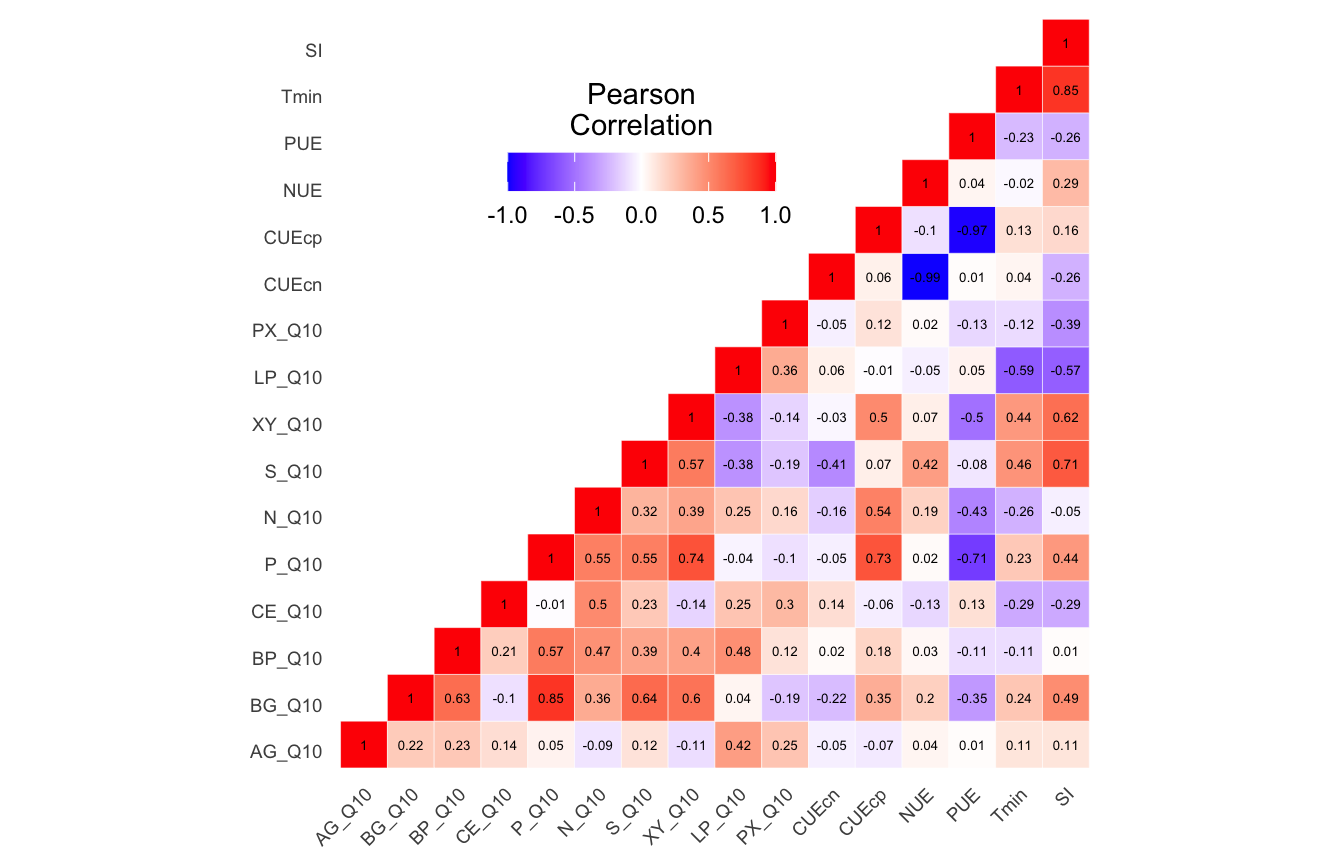
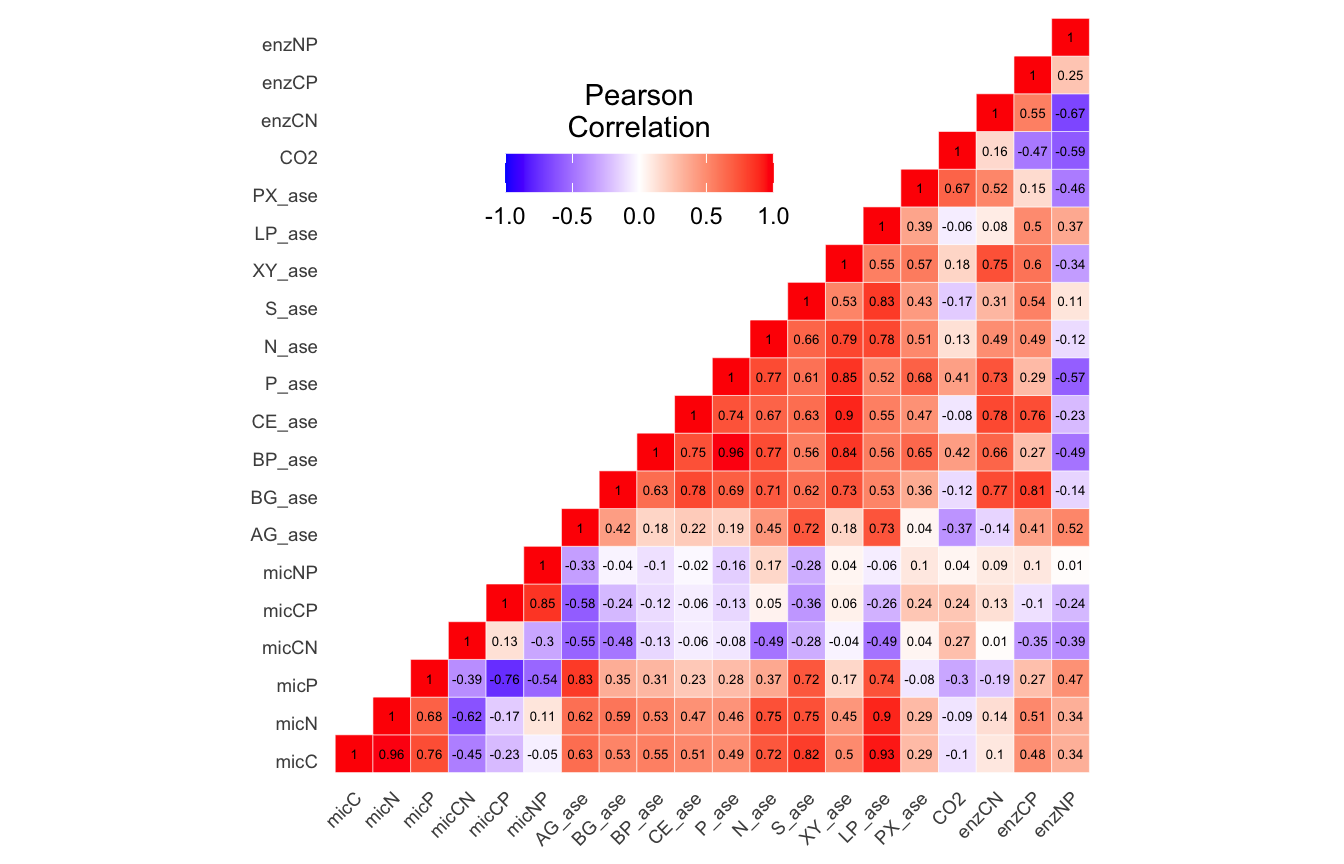
(ITS) Table 2 | Results of the Shapiro-Wilk Normality Tests. P-values in red are significance (p-value < 0.05) meaning the parameter needs to be normalized.
Looks like we need to transform 21 metadata parameters.
Normalize Parameters
Here we use the R package bestNormalize to find and execute the best normalizing transformation. The function will test the following normalizing transformations:
arcsinh_x performs an arcsinh transformation.
boxcox Perform a Box-Cox transformation and center/scale a vector to attempt normalization. boxcox estimates the optimal value of lambda for the Box-Cox transformation. The function will return an error if a user attempt to transform nonpositive data.
yeojohnson Perform a Yeo-Johnson Transformation and center/scale a vector to attempt normalization. yeojohnson estimates the optimal value of lambda for the Yeo-Johnson transformation. The Yeo-Johnson is similar to the Box-Cox method, however it allows for the transformation of nonpositive data as well.
orderNorm The Ordered Quantile (ORQ) normalization transformation, orderNorm(), is a rank-based procedure by which the values of a vector are mapped to their percentile, which is then mapped to the same percentile of the normal distribution. Without the presence of ties, this essentially guarantees that the transformation leads to a uniform distribution.
log_x performs a simple log transformation. The parameter a is essentially estimated by the training set by default (estimated as the minimum possible to some extent epsilon), while the base must be specified beforehand. The default base of the log is 10.
sqrt_x performs a simple square-root transformation. The parameter a is essentially estimated by the training set by default (estimated as the minimum possible), while the base must be specified beforehand.
exp_x performs a simple exponential transformation.
See this GitHub issue (#5) for a description on getting reproducible results. Apparently, you can get different results because the bestNormalize() function uses repeated cross-validation (and doesn’t automatically set the seed), so the results will be slightly different each time the function is executed.
Show the code
set.seed(119)for(iinmd_to_tranform){tmp_md<-its18_select_mc$map_loadedtmp_best_norm<-bestNormalize(tmp_md[[i]], r =1, k =5, loo =TRUE)tmp_name<-purrr::map_chr(i, ~paste0(., "_best_norm_test"))assign(tmp_name, tmp_best_norm)print(tmp_name)rm(list =ls(pattern ="tmp_"))}
Show the chosen transformations
## bestNormalize Chosen transformation of AST ##
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
25.80 26.26 28.80 29.90 41.77
_____________________________________
## bestNormalize Chosen transformation of H2O ##
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
0.204 0.261 0.377 0.385 0.401
_____________________________________
## bestNormalize Chosen transformation of Al ##
Standardized asinh(x) Transformation with 13 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 0.005384372
- sd (before standardization) = 0.007762057
_____________________________________
## bestNormalize Chosen transformation of Fe ##
Standardized asinh(x) Transformation with 13 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 0.01076821
- sd (before standardization) = 0.0103749
_____________________________________
## bestNormalize Chosen transformation of TEB ##
orderNorm Transformation with 13 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
41.6 43.3 45.1 48.7 61.7
_____________________________________
## bestNormalize Chosen transformation of ECEC ##
orderNorm Transformation with 13 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
41.7 43.3 45.3 48.8 61.8
_____________________________________
## bestNormalize Chosen transformation of minNO3 ##
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
11.31 19.67 22.71 37.33 106.42
_____________________________________
## bestNormalize Chosen transformation of minTIN ##
orderNorm Transformation with 13 nonmissing obs and ties
- 12 unique values
- Original quantiles:
0% 25% 50% 75% 100%
12.96 21.50 23.99 42.08 110.75
_____________________________________
## bestNormalize Chosen transformation of DOC ##
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
33.20 44.30 56.68 63.79 118.74
_____________________________________
## bestNormalize Chosen transformation of BG_ase ##
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
3.04 3.92 4.22 4.62 9.09
_____________________________________
## bestNormalize Chosen transformation of BP_ase ##
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
1.77 2.33 3.02 3.50 7.28
_____________________________________
## bestNormalize Chosen transformation of P_ase ##
Standardized Box Cox Transformation with 13 nonmissing obs.:
Estimated statistics:
- lambda = -0.9999576
- mean (before standardization) = 0.9480149
- sd (before standardization) = 0.01593697
_____________________________________
## bestNormalize Chosen transformation of N_ase ##
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
2.57 2.95 3.58 4.52 8.34
_____________________________________
## bestNormalize Chosen transformation of XY_ase ##
orderNorm Transformation with 13 nonmissing obs and ties
- 11 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.66 0.76 1.11 1.36 2.52
_____________________________________
## bestNormalize Chosen transformation of BG_Q10 ##
orderNorm Transformation with 13 nonmissing obs and ties
- 11 unique values
- Original quantiles:
0% 25% 50% 75% 100%
1.31 1.36 1.39 1.56 1.76
_____________________________________
## bestNormalize Chosen transformation of CE_Q10 ##
orderNorm Transformation with 13 nonmissing obs and ties
- 12 unique values
- Original quantiles:
0% 25% 50% 75% 100%
1.76 1.87 1.95 2.00 2.36
_____________________________________
## bestNormalize Chosen transformation of CO2 ##
Standardized asinh(x) Transformation with 13 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 2.626573
- sd (before standardization) = 0.7396036
_____________________________________
## bestNormalize Chosen transformation of PX_ase ##
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
81.25 94.13 119.75 211.18 294.39
_____________________________________
## bestNormalize Chosen transformation of PX_Q10 ##
Standardized Yeo-Johnson Transformation with 13 nonmissing obs.:
Estimated statistics:
- lambda = -4.999963
- mean (before standardization) = 0.1968145
- sd (before standardization) = 0.001394949
_____________________________________
## bestNormalize Chosen transformation of CUEcp ##
orderNorm Transformation with 13 nonmissing obs and ties
- 9 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.15 0.17 0.19 0.21 0.33
_____________________________________
## bestNormalize Chosen transformation of PUE ##
orderNorm Transformation with 13 nonmissing obs and ties
- 6 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.77 0.88 0.90 0.91 0.92
_____________________________________
Show the complete bestNormalize results
## Results of bestNormalize for AST ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 3.2051
- Box-Cox: 3.2051
- Center+scale: 4.1282
- Exp(x): 17.9744
- Log_b(x+a): 3.2051
- orderNorm (ORQ): 0.1282
- sqrt(x + a): 4.1282
- Yeo-Johnson: 2.2821
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
25.80 26.26 28.80 29.90 41.77
_____________________________________
## Results of bestNormalize for H2O ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 7.2051
- Box-Cox: 6.2821
- Center+scale: 8.1282
- Exp(x): 6.2821
- Log_b(x+a): 7.2051
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 7.2051
- Yeo-Johnson: 6.2821
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
0.204 0.261 0.377 0.385 0.401
_____________________________________
## Results of bestNormalize for Al ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 7.5128
- Center+scale: 7.5128
- Exp(x): 7.5128
- Log_b(x+a): 9.359
- orderNorm (ORQ): 7.5128
- sqrt(x + a): 7.5128
- Yeo-Johnson: 7.5128
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized asinh(x) Transformation with 13 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 0.005384372
- sd (before standardization) = 0.007762057
_____________________________________
## Results of bestNormalize for Fe ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 7.2051
- Center+scale: 7.2051
- Exp(x): 7.2051
- Log_b(x+a): 7.2051
- orderNorm (ORQ): 7.2051
- sqrt(x + a): 7.2051
- Yeo-Johnson: 7.2051
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized asinh(x) Transformation with 13 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 0.01076821
- sd (before standardization) = 0.0103749
_____________________________________
## Results of bestNormalize for TEB ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.6667
- Box-Cox: 1.6667
- Center+scale: 4.7436
- Exp(x): 17.9744
- Log_b(x+a): 1.6667
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 3.2051
- Yeo-Johnson: 1.359
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
41.6 43.3 45.1 48.7 61.7
_____________________________________
## Results of bestNormalize for ECEC ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.6667
- Box-Cox: 1.6667
- Center+scale: 3.2051
- Exp(x): 17.9744
- Log_b(x+a): 1.6667
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 3.2051
- Yeo-Johnson: 1.359
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and ties
- 10 unique values
- Original quantiles:
0% 25% 50% 75% 100%
41.7 43.3 45.3 48.8 61.8
_____________________________________
## Results of bestNormalize for minNO3 ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 3.5128
- Box-Cox: 3.5128
- Center+scale: 5.359
- Exp(x): 17.9744
- Log_b(x+a): 3.5128
- orderNorm (ORQ): 0.7436
- sqrt(x + a): 2.5897
- Yeo-Johnson: 3.5128
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
11.31 19.67 22.71 37.33 106.42
_____________________________________
## Results of bestNormalize for minTIN ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.359
- Box-Cox: 2.2821
- Center+scale: 4.1282
- Exp(x): 17.9744
- Log_b(x+a): 1.359
- orderNorm (ORQ): 0.7436
- sqrt(x + a): 2.2821
- Yeo-Johnson: 2.2821
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and ties
- 12 unique values
- Original quantiles:
0% 25% 50% 75% 100%
12.96 21.50 23.99 42.08 110.75
_____________________________________
## Results of bestNormalize for DOC ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 2.8974
- Box-Cox: 2.8974
- Center+scale: 3.5128
- Exp(x): 17.9744
- Log_b(x+a): 2.8974
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 1.9744
- Yeo-Johnson: 2.8974
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
33.20 44.30 56.68 63.79 118.74
_____________________________________
## Results of bestNormalize for BG_ase ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 3.8205
- Box-Cox: 2.2821
- Center+scale: 4.1282
- Exp(x): 14.8974
- Log_b(x+a): 3.8205
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 3.5128
- Yeo-Johnson: 1.6667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
3.04 3.92 4.22 4.62 9.09
_____________________________________
## Results of bestNormalize for BP_ase ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.6667
- Box-Cox: 1.6667
- Center+scale: 4.1282
- Exp(x): 5.6667
- Log_b(x+a): 1.6667
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 1.6667
- Yeo-Johnson: 1.6667
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
1.77 2.33 3.02 3.50 7.28
_____________________________________
## Results of bestNormalize for P_ase ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.6667
- Box-Cox: 0.4359
- Center+scale: 3.2051
- Exp(x): 17.9744
- Log_b(x+a): 1.6667
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 1.9744
- Yeo-Johnson: 0.4359
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized Box Cox Transformation with 13 nonmissing obs.:
Estimated statistics:
- lambda = -0.9999576
- mean (before standardization) = 0.9480149
- sd (before standardization) = 0.01593697
_____________________________________
## Results of bestNormalize for N_ase ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.9744
- Box-Cox: 2.2821
- Center+scale: 2.8974
- Exp(x): 17.9744
- Log_b(x+a): 1.9744
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 2.8974
- Yeo-Johnson: 0.4359
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
2.57 2.95 3.58 4.52 8.34
_____________________________________
## Results of bestNormalize for XY_ase ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.0513
- Box-Cox: 0.4359
- Center+scale: 2.8974
- Exp(x): 9.0513
- Log_b(x+a): 1.0513
- orderNorm (ORQ): 0.1282
- sqrt(x + a): 1.359
- Yeo-Johnson: 0.4359
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and ties
- 11 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.66 0.76 1.11 1.36 2.52
_____________________________________
## Results of bestNormalize for BG_Q10 ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.9744
- Box-Cox: 1.9744
- Center+scale: 2.2821
- Exp(x): 5.359
- Log_b(x+a): 1.9744
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 1.9744
- Yeo-Johnson: 1.9744
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and ties
- 11 unique values
- Original quantiles:
0% 25% 50% 75% 100%
1.31 1.36 1.39 1.56 1.76
_____________________________________
## Results of bestNormalize for CE_Q10 ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.6667
- Box-Cox: 1.6667
- Center+scale: 1.6667
- Exp(x): 4.1282
- Log_b(x+a): 1.6667
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 1.6667
- Yeo-Johnson: 0.7436
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and ties
- 12 unique values
- Original quantiles:
0% 25% 50% 75% 100%
1.76 1.87 1.95 2.00 2.36
_____________________________________
## Results of bestNormalize for CO2 ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.0513
- Box-Cox: 3.5128
- Center+scale: 11.5128
- Exp(x): 17.9744
- Log_b(x+a): 1.0513
- orderNorm (ORQ): 1.0513
- sqrt(x + a): 2.8974
- Yeo-Johnson: 1.9744
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized asinh(x) Transformation with 13 nonmissing obs.:
Relevant statistics:
- mean (before standardization) = 2.626573
- sd (before standardization) = 0.7396036
_____________________________________
## Results of bestNormalize for PX_ase ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 1.9744
- Box-Cox: 2.2821
- Center+scale: 4.1282
- Exp(x): 17.9744
- Log_b(x+a): 1.9744
- orderNorm (ORQ): 0.4359
- sqrt(x + a): 1.9744
- Yeo-Johnson: 2.2821
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and no ties
- Original quantiles:
0% 25% 50% 75% 100%
81.25 94.13 119.75 211.18 294.39
_____________________________________
## Results of bestNormalize for PX_Q10 ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 14.5897
- Box-Cox: 3.2051
- Center+scale: 17.9744
- Exp(x): 17.9744
- Log_b(x+a): 11.5128
- orderNorm (ORQ): 2.2821
- sqrt(x + a): 17.9744
- Yeo-Johnson: 1.0513
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
Standardized Yeo-Johnson Transformation with 13 nonmissing obs.:
Estimated statistics:
- lambda = -4.999963
- mean (before standardization) = 0.1968145
- sd (before standardization) = 0.001394949
_____________________________________
## Results of bestNormalize for CUEcp ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 3.5128
- Box-Cox: 1.359
- Center+scale: 3.5128
- Exp(x): 4.4359
- Log_b(x+a): 1.359
- orderNorm (ORQ): 0.7436
- sqrt(x + a): 3.5128
- Yeo-Johnson: 2.5897
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and ties
- 9 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.15 0.17 0.19 0.21 0.33
_____________________________________
## Results of bestNormalize for PUE ##
Best Normalizing transformation with 13 Observations
Estimated Normality Statistics (Pearson P / df, lower => more normal):
- arcsinh(x): 5.9744
- Box-Cox: 7.2051
- Center+scale: 5.9744
- Exp(x): 5.9744
- Log_b(x+a): 7.2051
- orderNorm (ORQ): 2.5897
- sqrt(x + a): 5.9744
- Yeo-Johnson: 5.9744
Estimation method: Out-of-sample via leave-one-out CV
Based off these, bestNormalize chose:
orderNorm Transformation with 13 nonmissing obs and ties
- 6 unique values
- Original quantiles:
0% 25% 50% 75% 100%
0.77 0.88 0.90 0.91 0.92
_____________________________________
Great, now we can add the normalized transformed data back to our mctoolsr metadata file.
Ok. Looks like bestNormalize was unable to find a suitable transformation for Al and Fe. This is likely because there is very little variation in these metadata and/or there are too few significant digits.
Normalized Metadata
Finally, here is a new summary table that includes all of the normalized data.
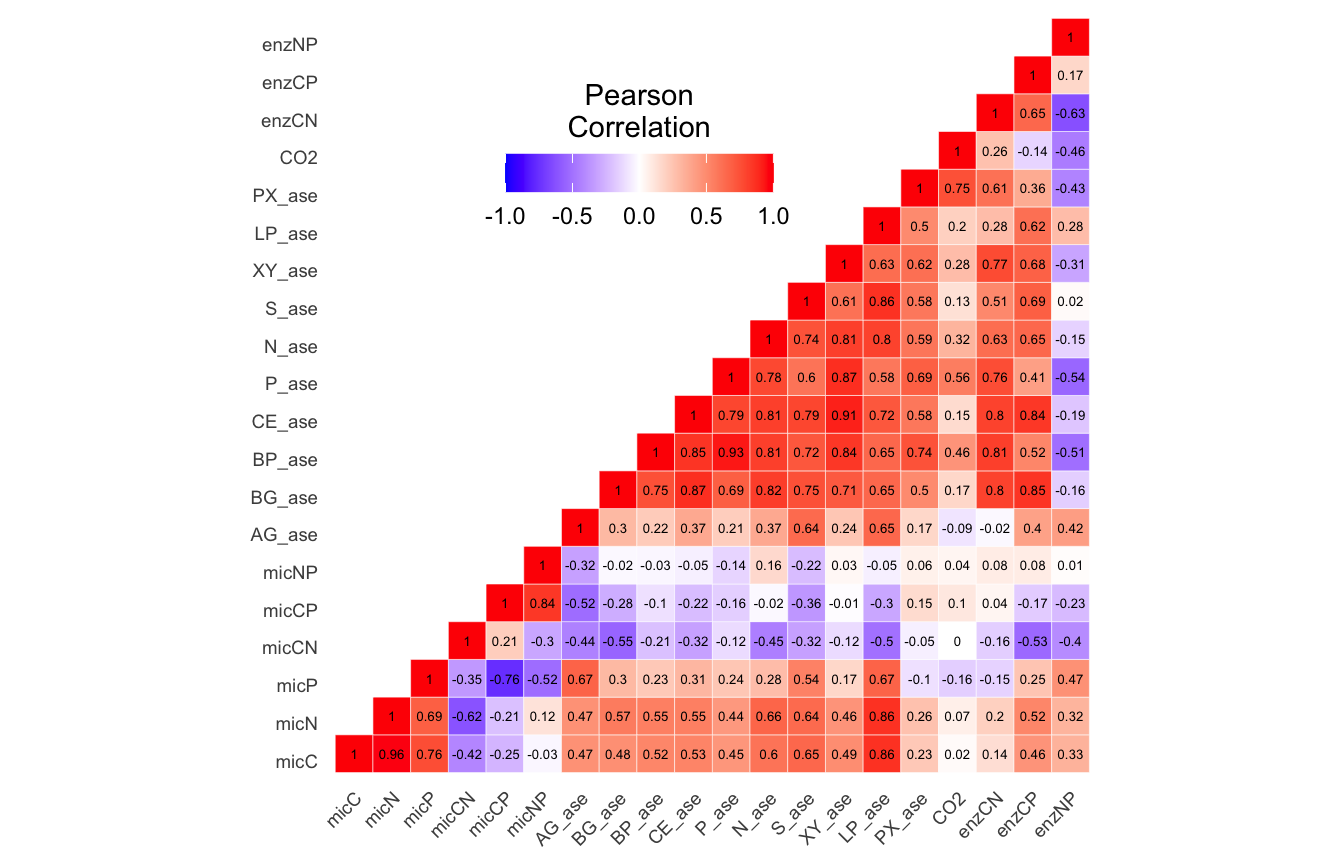
(ITS) Table 3 | Results of bestNormalize function applied to each parameter.
Autocorrelation Tests
Next, we test the metadata for autocorrelations. Do we do this on the original data or the transformed data? No idea, so let’s do both.
Let’s see if any on the metadata groups are significantly correlated with the community data. Basically, we create distance matrices for the community data and each metadata group and then run Mantel tests for all comparisons. For the community data we calculate Bray-Curtis distances for the community data and Euclidean distances for the metadata. We use the function mantel.test from the ape package and mantel from the vegan package for the analyses.
In summary, we test both mantel.test and mantel on Bray-Curtis distance community distances against Euclidean distances for each metadata group (edaphic, soil_funct, temp_adapt) a) before normalizing and before removing autocorrelated parameters, b) before normalizing and after removing autocorrelated parameters, c) after normalizing and before removing autocorrelated parameters, and d) after normalizing and after removing autocorrelated parameters.
(ITS) Table 4 | Summary of Dissimilarity Correlation Tests using mantel.test from the ape package and mantel from the vegan. P-values in red indicate significance (p-value < 0.05)
Moving on.
Best Subset of Variables
Now we want to know which of the metadata parameters are the most strongly correlated with the community data. For this we use the bioenv function from the vegan package. bioenv—Best Subset of Environmental Variables with Maximum (Rank) Correlation with Community Dissimilarities—finds the best subset of environmental variables, so that the Euclidean distances of scaled environmental variables have the maximum (rank) correlation with community dissimilarities.
Since we know that each of the Mantel tests we ran above are significant, here we will use the metadata set where autocorrelated parameters were removed and the remainder of the parameters were normalized (where applicable based on the Shapiro tests).
We run bioenv against the three groups of metadata parameters. We then run bioenv again, but this time against the individual parameters identified as significantly correlated.
Call:
bioenv(comm = wisconsin(tmp_comm), env = tmp_env, method = "spearman", index = "bray", upto = ncol(tmp_env), metric = "euclidean")
Subset of environmental variables with best correlation to community data.
Correlations: spearman
Dissimilarities: bray
Metric: euclidean
Best model has 1 parameters (max. 15 allowed):
AST
with correlation 0.65665
Show the results of individual edaphic metadata Mantel tests
$AST
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 1
Significance: 0.001
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.192 0.242 0.272 0.339
Permutation: free
Number of permutations: 999
bioenv found the following edaphic properties significantly correlated with the community data: AST
Call:
bioenv(comm = wisconsin(tmp_comm), env = tmp_env, method = "spearman", index = "bray", upto = ncol(tmp_env), metric = "euclidean")
Subset of environmental variables with best correlation to community data.
Correlations: spearman
Dissimilarities: bray
Metric: euclidean
Best model has 3 parameters (max. 11 allowed):
XY_ase PX_ase enzNP
with correlation 0.7491622
Show the results of individual functional response metadata Mantel tests
$enzNP
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.5529
Significance: 0.001
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.202 0.277 0.330 0.417
Permutation: free
Number of permutations: 999
$PX_ase
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.6854
Significance: 0.001
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.209 0.282 0.361 0.412
Permutation: free
Number of permutations: 999
$XY_ase
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.5054
Significance: 0.002
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.202 0.261 0.308 0.359
Permutation: free
Number of permutations: 999
bioenv found the following soil functions significantly correlated with the community data: enzNP, PX_ase, XY_ase
Call:
bioenv(comm = wisconsin(tmp_comm), env = tmp_env, method = "spearman", index = "bray", upto = ncol(tmp_env), metric = "euclidean")
Subset of environmental variables with best correlation to community data.
Correlations: spearman
Dissimilarities: bray
Metric: euclidean
Best model has 2 parameters (max. 12 allowed):
XY_Q10 Tmin
with correlation 0.5637527
Show the results of individual temperature adaptation metadata Mantel tests
$XY_Q10
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.7258
Significance: 0.001
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.134 0.200 0.241 0.316
Permutation: free
Number of permutations: 999
$Tmin
Mantel statistic based on Spearman's rank correlation rho
Call:
mantel(xdis = bioenv_best, ydis = tmp_md, method = "spearman", permutations = 999)
Mantel statistic r: 0.616
Significance: 0.001
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.189 0.245 0.297 0.360
Permutation: free
Number of permutations: 999
bioenv found the following temperature adaptations significantly correlated with the community data: XY_Q10, Tmin
Distance-based Redundancy
Now we turn our attention to distance-based redundancy analysis (dbRDA), an ordination method similar to Redundancy Analysis (rda) but it allows non-Euclidean dissimilarity indices, such as Manhattan or Bray–Curtis distance. For this, we use capscale from the vegan package. capscale is a constrained versions of metric scaling (principal coordinates analysis), which are based on the Euclidean distance but can be used, and are more useful, with other dissimilarity measures. The functions can also perform unconstrained principal coordinates analysis, optionally using extended dissimilarities.
For each of the three metadata subsets, we perform the following steps:
Run rankindex to compare metadata and community dissimilarity indices for gradient detection. This will help us select the best dissimilarity metric to use.
Run capscale for distance-based redundancy analysis.
Run envfit to fit environmental parameters onto the ordination. This function basically calculates correlation scores between the metadata parameters and the ordination axes.
Select metadata parameters significant for bioenv (see above) and/or envfit analyses.
euc man gow bra kul
0.15398526 0.29983940 -0.02940098 0.29983940 0.29983940
Let’s run capscale using Bray-Curtis. Note, we have 15 metadata parameters in this group but, for some reason, capscale only works with 13 parameters. This may have to do with degrees of freedom?
Call: capscale(formula = tmp_comm ~ AST + H2O + N + P + ECEC + pH + NH4 +
NO3 + PO4 + DOC + DOCN, data = tmp_md, distance = "bray")
Inertia Proportion Rank
Total 3.88624 1.00000
Constrained 3.66503 0.94308 11
Unconstrained 0.22120 0.05692 1
Inertia is squared Bray distance
-- NOTE:
Species scores projected from 'tmp_comm'
Eigenvalues for constrained axes:
CAP1 CAP2 CAP3 CAP4 CAP5 CAP6 CAP7 CAP8 CAP9 CAP10 CAP11
0.9655 0.5791 0.4327 0.3671 0.2987 0.2608 0.2301 0.1867 0.1339 0.1135 0.0970
Eigenvalues for unconstrained axes:
MDS1
0.2212
Now we can look at the variance against each principal component.
And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The ggplot function autoplot stores these data in a more accessible way than the raw results from capscale
Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function scores which gets species or site scores from the ordination.
Show the code
tmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")edaphic_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")
Now we have a new data frame that contains sample details and capscale values.
SampleID PLOT TREAT TREAT_T PAIR CAP1 CAP2
1 P10_D00_010_C0E P10 C 0 E -0.67150210 -1.07183211
2 P02_D00_010_C0A P02 C 0 A -0.91911652 -0.41647565
3 P04_D00_010_C0B P04 C 0 B -0.95654588 0.64087450
4 P06_D00_010_C0C P06 C 0 C -0.87992057 0.68082267
5 P08_D00_010_C0D P08 C 0 D -0.07766291 -0.98166349
6 P01_D00_010_W3A P01 W 3 A -0.79165161 -0.43340347
7 P03_D00_010_W3B P03 W 3 B 0.24722812 0.81805851
8 P07_D00_010_W3D P07 W 3 D 0.20151965 1.07639049
9 P09_D00_010_W3E P09 W 3 E 0.58233467 0.46562279
10 P01_D00_010_W8A P01 W 8 A 0.92158009 -0.06825914
11 P03_D00_010_W8B P03 W 8 B 0.94079184 -0.52231363
12 P05_D00_010_W8C P05 W 8 C 0.87642984 -0.89155825
13 P07_D00_010_W8D P07 W 8 D 0.52651539 0.70373678
We can then do the same with the metadata vectors. Here though we only need the scores and parameter name.
envfit found that AST, DOC were significantly correlated.
Now let’s see if the same parameters are significant for the envfit and bioenv analyses.
[1] "Significant parameters from bioenv analysis."
[1] "AST"
_____________________________________
[1] "Significant parameters from envfit analysis."
[1] "AST" "DOC"
_____________________________________
[1] "Found in bioenv but not envfit."
character(0)
_____________________________________
[1] "Found in envfit but not bioenv."
[1] "DOC"
_____________________________________
[1] "Found in envfit and bioenv."
[1] "AST" "DOC"
euc man gow bra kul
0.06025620 0.38790324 -0.06200129 0.38790324 0.38790324
Let’s run capscale using Bray-Curtis Note, we have 11 metadata parameters in this group but, for some reason, capscale only works with 13 parameters. This may have to do with degrees of freedom?
Now we can look at the variance against each principal component.
And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The ggplot function autoplot stores these data in a more accessible way than the raw results from capscale
Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function scores which gets species or site scores from the ordination.
Show the code
tmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")soil_funct_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")
Now we have a new data frame that contains sample details and capscale values.
SampleID PLOT TREAT TREAT_T PAIR CAP1 CAP2
1 P10_D00_010_C0E P10 C 0 E -0.69489162 -0.6675049
2 P02_D00_010_C0A P02 C 0 A -0.91653485 -0.6229848
3 P04_D00_010_C0B P04 C 0 B -0.95221897 0.3986135
4 P06_D00_010_C0C P06 C 0 C -0.85713972 0.6268677
5 P08_D00_010_C0D P08 C 0 D -0.09115803 -1.0802252
6 P01_D00_010_W3A P01 W 3 A -0.80868973 -0.1643386
7 P03_D00_010_W3B P03 W 3 B 0.27067347 0.9200087
8 P07_D00_010_W3D P07 W 3 D 0.20427135 1.0264780
9 P09_D00_010_W3E P09 W 3 E 0.57963547 0.4440199
10 P01_D00_010_W8A P01 W 8 A 0.91663981 -0.1382072
11 P03_D00_010_W8B P03 W 8 B 0.92199435 -0.6783194
12 P05_D00_010_W8C P05 W 8 C 0.86406950 -0.9575454
13 P07_D00_010_W8D P07 W 8 D 0.56334898 0.8931377
We can then do the same with the metadata vectors. Here though we only need the scores and parameter name.
euc man gow bra kul
0.05132842 0.39465598 0.27457353 0.39465598 0.39465598
Let’s run capscale using Bray-Curtis. Note, we have 12 metadata parameters in this group but, for some reason, capscale only works with 13 parameters. This may have to do with degrees of freedom?
Now we can look at the variance against each principal component.
And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The ggplot function autoplot stores these data in a more accessible way than the raw results from capscale
Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function scores which gets species or site scores from the ordination.
Show the code
tmp_samp_scores<-dplyr::filter(tmp_auto_plt$plot_env$obj, Score=="sites")tmp_samp_scores[,1]<-NULLtmp_samp_scores<-tmp_samp_scores%>%dplyr::rename(SampleID =Label)tmp_md_sub<-tmp_md[, 1:4]tmp_md_sub<-tmp_md_sub%>%tibble::rownames_to_column("SampleID")temp_adapt_plot_data<-dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")
Now we have a new data frame that contains sample details and capscale values.
SampleID PLOT TREAT TREAT_T PAIR CAP1 CAP2
1 P10_D00_010_C0E P10 C 0 E -0.70909404 0.8884734
2 P02_D00_010_C0A P02 C 0 A -0.88932428 0.4678685
3 P04_D00_010_C0B P04 C 0 B -0.94720017 -0.5168746
4 P06_D00_010_C0C P06 C 0 C -0.94117253 -0.7283971
5 P08_D00_010_C0D P08 C 0 D -0.09697067 1.0013016
6 P01_D00_010_W3A P01 W 3 A -0.79138504 0.3434287
7 P03_D00_010_W3B P03 W 3 B 0.28447425 -0.8800769
8 P07_D00_010_W3D P07 W 3 D 0.18992920 -0.9828048
9 P09_D00_010_W3E P09 W 3 E 0.57704340 -0.4016378
10 P01_D00_010_W8A P01 W 8 A 0.98434765 0.1432237
11 P03_D00_010_W8B P03 W 8 B 0.88625427 0.6330378
12 P05_D00_010_W8C P05 W 8 C 0.84253827 0.9056297
13 P07_D00_010_W8D P07 W 8 D 0.61055970 -0.8731722
We can then do the same with the metadata vectors. Here though we only need the scores and parameter name.
envfit found that XY_Q10, CUEcp, Tmin were significantly correlated.
Now let’s see if the same parameters are significant for the envfit and bioenv analyses.
[1] "Significant parameters from bioenv analysis."
[1] "XY_Q10" "Tmin"
_____________________________________
[1] "Significant parameters from envfit analysis."
[1] "XY_Q10" "CUEcp" "Tmin"
_____________________________________
[1] "Found in bioenv but not envfit."
character(0)
_____________________________________
[1] "Found in envfit but not bioenv."
[1] "CUEcp"
_____________________________________
[1] "Found in envfit and bioenv."
[1] "XY_Q10" "CUEcp" "Tmin"
(ITS) Figure 7 | Distance-based Redundancy Analysis (db-RDA) of PIME filtered data based on Bray-Curtis dissimilarity showing the relationships between community composition change versus edaphic properties.
(ITS) Figure 8 | Distance-based Redundancy Analysis (db-RDA) of PIME filtered data based on Bray-Curtis dissimilarity showing the relationships between community composition change versus microbial functional response.
(ITS) Figure 9 | Distance-based Redundancy Analysis (db-RDA) of PIME filtered data based on Bray-Curtis dissimilarity showing the relationships between community composition change versus temperature adaptation.
Workflow Output
Data products generated in this workflow can be downloaded from figshare.
The source code for this page can be accessed on GitHub by clicking this link.
Data Availability
Data generated in this workflow and the Rdata need to run the workflow can be accessed on figshare at 10.25573/data.16828294.
Last updated on
[1] "2025-08-23 13:49:51 PDT"
References
Oksanen, Jari, F. Guillaume Blanchet, Roeland Kindt, Pierre Legendre, Peter R. Minchin, R. B. O’Hara, Gavin L. Simpson, Peter Sólymos, M. Henry H. Stevens, and Helene Wagner. 2012. “Vegan: Community Ecology Package.”R Package Version 2 (0). https://cran.r-project.org/web/packages/vegan/index.html.
Source Code
---title: "8. Metadata Analysis"description: | Workflow to assess the relationship between environmental parameters and microbial communities. ---```{r}#| message: false#| results: hide#| code-fold: true#| code-summary: "Click here for setup information"knitr::opts_chunk$set(eval =FALSE, echo =TRUE)set.seed(119)#library(conflicted)#pacman::p_depends(vegan, local = TRUE) #pacman::p_depends_reverse(vegan, local = TRUE) library(phyloseq); packageVersion("phyloseq")pacman::p_load(ape, bestNormalize, tidyverse, gdata, mctoolsr, ggpubr, ggvegan, ggfortify, vegan, reactable, downloadthis, captioner, ggplot2,install =FALSE, update =FALSE)options(scipen=999)knitr::opts_current$get(c("cache","cache.path","cache.rebuild","dependson","autodep"))``````{r}#| include: false#| eval: true#| comment: "Load for summary #3"remove(list =ls())load("page_build/metadata_summary.rdata")objects()``````{r}#| echo: false#| eval: true#| results: hide#| comment: "Create the caption(s) with captioner"caption_tab_ssu <-captioner(prefix ="(16S rRNA) Table", suffix =" |", style ="b")caption_fig_ssu <-captioner(prefix ="(16S rRNA) Figure", suffix =" |", style ="b")caption_tab_its <-captioner(prefix ="(ITS) Table", suffix =" |", style ="b")caption_fig_its <-captioner(prefix ="(ITS) Figure", suffix =" |", style ="b")caption_tab <-captioner(prefix ="(common) Table", suffix =" |", style ="b")caption_fig <-captioner(prefix ="(common) Figure", suffix =" |", style ="b")# Create a function for referring to the tables in textref <-function(x) str_extract(x, "[^|]*") %>%trimws(which ="right", whitespace ="[ ]")``````{r}#| echo: false#| eval: truesource("assets/captions/captions_metadata.R")```# SynopsisIn this workflow we compare the environmental metadata with the microbial community data. Part 1 involves the [16S rRNA](#s-rrna-1) community data and Part 2 deals with the [ITS](#its-1) community data. Each workflow contains the same major steps:1) Metadata Normality Tests: Shapiro-Wilk Normality Test to test whether each matadata parameter is normally distributed.2) Normalize Parameters: R package [`bestNormalize`](https://github.com/petersonR/bestNormalize) to find and execute the best normalizing transformation. 3) Split Metadata parameters into groups: a) Environmental and edaphic properties, b) Microbial functional responses, and c) Temperature adaptation properties.4) Autocorrelation Tests: Test all possible pair-wise comparisons, on both normalized and non-normalized data sets, for each group.5) Remove autocorrelated parameters from each group.6) Dissimilarity Correlation Tests: Use Mantel Tests to see if any on the metadata groups are significantly correlated with the community data.7) Best Subset of Variables: Determine which of the metadata parameters from each group are the most strongly correlated with the community data. For this we use the `bioenv` function from the `vegan`[@oksanen2013community] package.8) Distance-based Redundancy Analysis: Ordination analysis of samples and metadata vector overlays using `capscale`, also from the `vegan` package. ## Workflow InputFiles needed to run this workflow can be downloaded from figshare. You will also need the metadata table, which can be accessed below. <iframe src="https://widgets.figshare.com/articles/16826779/embed?show_title=1" width="100%" height="251" allowfullscreen frameborder="0"></iframe>## Metadata In this section we provide access to the complete environmental metadata containing 61 parameters across all 15 samples. <br/>```{r}#| echo: false#| eval: truemetad <-read.table("files/metadata/tables/metadata.txt",header =TRUE, sep ="\t")seq_table <- metad ```<small>`r caption_tab("metadata_results")`</small>```{r}#| echo: false#| eval: truereactable(seq_table,defaultColDef =colDef(headerStyle =list(fontSize ="0.8em"),header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =1),align ="center", filterable =TRUE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold") ), columns =list(Sample_ID =colDef(name ="Sample ID", sticky ="left", style =list(borderRight ="1px solid #eee"),headerStyle =list(borderRight ="1px solid #eee", fontSize ="0.9em"), align ="left",minWidth =150), raw_reads =colDef(footer =function(values) sprintf("%.0f", sum(values))) ), searchable =TRUE, defaultPageSize =5, pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =FALSE, wrap =FALSE, showSortable =TRUE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```{{< downloadthis files/metadata/tables/metadata.txt dname="metadata" label="Download table as text file" icon="table" type="primary" >}}# Multivariate SummarySummary results for of `envfit` and `bioenv` tests for edaphic properties, soil functional response, and temperature adaptation for both 16S rRNA and ITS data sets.## 16S rRNAMetadata parameters removed based on autocorrelation results.- edaphic properties: TEB + DON + Na + Al + Ca- soil functional response: micN + micNP + enzCN + enzCP + BP~ase~ + CE~ase~ + LP~ase~ + N~ase~ + P~ase~- temperature adaptation: NUE + PUE + SIMetadata parameters removed from capscale analysis based on Degrees of Freedom.- edaphic properties: Mg + Mn- soil functional response: NONE- temperature adaptation: NONE<br/>```{r}#| echo: false#| eval: trueseq_table <-read.table("files/metadata/tables/ssu18_sig_envfit_bioenv_summary.txt", header =TRUE, sep ="\t")``````{r}#| echo: false#| eval: true#| warning: false#| message: falseseq_table <- ssu18_sig_summary %>% dplyr::rename("envfit_r2"=3, "envfit_pval"=4, "bioenv_r"=5, "bioenv_pval"=6)seq_table <- seq_table %>% dplyr::mutate(across(.cols =c(3:6), round, digits =3)) seq_table[is.na(seq_table)] <-""```<small>`r caption_tab_ssu("ssu_multi_summ")`</small>```{r}#| echo: false#| eval: truereactable(seq_table, groupBy ="metadata_set", defaultExpanded =TRUE,defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =3),align ="center", filterable =FALSE, sortable =FALSE, resizable =TRUE,footerStyle =list(fontWeight ="bold"), minWidth =50), columns =list(metadata_set =colDef(name ="Metadata group", sticky ="left", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee"), align ="left",minWidth =90),parameter =colDef(name ="parameter", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee")),envfit_r2 =colDef(name ="r2"),envfit_pval =colDef(name ="P(>r)", style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color, fontWeight ="bold", borderRight ="5px solid #eee")}, headerStyle =list(borderRight ="5px solid #eee")),bioenv_r =colDef(name ="r", minWidth =40),bioenv_pval =colDef(name ="p-value", style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color, fontWeight ="bold")})), columnGroups =list(colGroup(name ="envfit", columns =c("envfit_r2", "envfit_pval"), headerStyle =list(fontSize ="1.2em")),colGroup(name ="bioenv", columns =c("bioenv_r", "bioenv_pval"), headerStyle =list(fontSize ="1.2em"))),searchable =FALSE, defaultPageSize =15, showPagination =FALSE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =FALSE, showSortable =FALSE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```{{< downloadthis files/metadata/tables/ssu18_sig_envfit_bioenv_summary.txt dname="ssu_multivariate_summary" label="Download table as text file" icon="table" type="primary" >}}## ITSMetadata parameters removed based on autocorrelation results.- edaphic properties: TEB + DON + Na + Al + Ca- soil functional response: micN + micNP + enzCN + enzCP + BP~ase~ + CE~ase~ + LP~ase~ + N~ase~ + P~ase~- temperature adaptation: NUE + PUE + P~Q10~ + SIMetadata parameters removed from capscale analysis based on Degrees of Freedom.- edaphic properties: Mg + Mn + Na + Al + Fe + K- soil functional response: NONE- temperature adaptation: S~Q10~<br/>```{r}#| echo: false#| eval: trueseq_table <-read.table("files/metadata/tables/its18_sig_envfit_bioenv_summary.txt", header =TRUE, sep ="\t")``````{r}#| echo: false#| eval: trueseq_table <- its18_sig_summary %>% dplyr::rename("envfit_r2"=3, "envfit_pval"=4, "bioenv_r"=5, "bioenv_pval"=6)seq_table <- seq_table %>% dplyr::mutate(across(.cols =c(3:6), round, digits =3)) seq_table[is.na(seq_table)] <-""```<small>`r caption_tab_its("its_multi_summ")`</small>```{r}#| echo: false#| eval: truereactable(seq_table, groupBy ="metadata_set", defaultExpanded =TRUE,defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =3),align ="center", filterable =FALSE, sortable =FALSE, resizable =TRUE,footerStyle =list(fontWeight ="bold"), minWidth =50), columns =list(metadata_set =colDef(name ="Metadata group", sticky ="left", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee"), align ="left",minWidth =90),parameter =colDef(name ="parameter", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee")),envfit_r2 =colDef(name ="r2"),envfit_pval =colDef(name ="P(>r)", style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color, fontWeight ="bold", borderRight ="5px solid #eee")}, headerStyle =list(borderRight ="5px solid #eee")),bioenv_r =colDef(name ="r", minWidth =40),bioenv_pval =colDef(name ="p-value", style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color, fontWeight ="bold")})), columnGroups =list(colGroup(name ="envfit", columns =c("envfit_r2", "envfit_pval"), headerStyle =list(fontSize ="1.2em")),colGroup(name ="bioenv", columns =c("bioenv_r", "bioenv_pval"), headerStyle =list(fontSize ="1.2em"))),searchable =FALSE, defaultPageSize =15, showPagination =FALSE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =FALSE, showSortable =FALSE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```{{< downloadthis files/metadata/tables/its18_sig_envfit_bioenv_summary.txt dname="its_multivariate_summary" label="Download table as text file" icon="table" type="primary" >}}```{r}#| include: false#| eval: true#| comment: "Load to build page only #2"load("page_build/metadata_ssu18_wf.rdata")objects()``````{r}#| echo: falseswel_col <-c("#2271B2", "#71B222", "#B22271")tmp_1 <- ssu18_ps_work_mc$data_loadedtmp_2 <- ssu18_ps_pime_mc$data_loadedtmp_1 <- tmp_1 %>% tibble::rownames_to_column("ID")tmp_2 <- tmp_2 %>% tibble::rownames_to_column("ID")tmp_3 <-left_join(tmp_2, tmp_1, by ="ID")tmp_3 <- tmp_3[ , order(names(tmp_3))]write.table(tmp_3, "tmp_3.txt", sep ="\t", quote =FALSE)```# 16s rRNA## Data Formating ```{r}#| include: false#| comment: "Initial Load for ANALYSIS #1"set.seed(119)ssu18_ps_work <-readRDS("files/alpha/rdata/ssu18_ps_work.rds")ssu18_ps_filt <-readRDS("files/alpha/rdata/ssu18_ps_filt.rds")ssu18_ps_perfect <-readRDS("files/alpha/rdata/ssu18_ps_perfect.rds")ssu18_ps_pime <-readRDS("files/alpha/rdata/ssu18_ps_pime.rds")```1. Transform `ps` ObjectsThe first step is to transform the phyloseq objects. ```{r}#| code-fold: truesamp_ps <-c("ssu18_ps_work", "ssu18_ps_pime", "ssu18_ps_perfect", "ssu18_ps_work_otu", "ssu18_ps_pime_otu", "ssu18_ps_perfect_otu")for (i in samp_ps) { tmp_get <-get(i) tmp_ps <-transform_sample_counts(tmp_get, function(otu) 1e5* otu/sum(otu)) tmp_ps@phy_tree <-NULL tmp_ps <-prune_samples(sample_sums(tmp_ps) >0, tmp_ps) tmp_tree <-rtree(ntaxa(tmp_ps), rooted =TRUE, tip.label =taxa_names(tmp_ps)) tmp_samp <-data.frame(sample_data(tmp_get)) tmp_samp <- tmp_samp %>% dplyr::rename("TREAT_T"="TEMP") tmp_ps <-merge_phyloseq(tmp_ps, sample_data, tmp_tree)sample_data(tmp_ps) <- tmp_samp tmp_name <- purrr::map_chr(i, ~paste0(., "_prop"))print(tmp_name)assign(tmp_name, tmp_ps)rm(list =ls(pattern ="tmp_"))}```2. Format for `mctoolsr`Then a bit of formatting to make the data compatible with `mctoolsr`. ```{r}#| code-fold: truefor (i in samp_ps) { tmp_get <-get(purrr::map_chr(i, ~paste0(., "_prop"))) tmp_tax <-data.frame(tax_table(tmp_get)) tmp_tax$ASV_SEQ <-NULL tmp_col_names <-colnames(tmp_tax) tmp_tax_merge <- tmp_tax %>% tidyr::unite(taxonomy, all_of(tmp_col_names), sep =";") tmp_tax_merge <- tmp_tax_merge %>% tibble::rownames_to_column("#OTU_ID") tmp_otu <-data.frame(t(otu_table(tmp_get))) tmp_otu <- tmp_otu %>% tibble::rownames_to_column("#OTU_ID") tmp_otu_tax <- dplyr::left_join(tmp_otu, tmp_tax_merge, by ="#OTU_ID") tmp_samp <-data.frame(sample_data(tmp_get)) tmp_samp[,c(1,3)] <-NULL tmp_samp <- tmp_samp %>% tibble::rownames_to_column("#SampleID") tmp_metad <- metad %>% dplyr::rename("#SampleID"="id") tmp_metad[,2:5] <-NULL tmp_md <- dplyr::left_join(tmp_samp, tmp_metad, by ="#SampleID") tmp_otu_name <- purrr::map_chr(i, ~paste0(., "_otu_tax"))print(tmp_otu_name)assign(tmp_otu_name, tmp_otu_tax) tmp_md_name <- purrr::map_chr(i, ~paste0(., "_md"))print(tmp_md_name)assign(tmp_md_name, tmp_md) tmp_path <-file.path("files/metadata/tables/")write_delim(tmp_otu_tax, paste(tmp_path, tmp_otu_name, ".txt", sep =""), delim ="\t")write_delim(tmp_md, paste(tmp_path, tmp_md_name, ".txt", sep =""), delim ="\t")rm(list =ls(pattern ="tmp_"))}``````{r}#| code-fold: truefor (i in samp_ps) { tmp_path <-file.path("files/metadata/tables/") tmp_otu_name <- purrr::map_chr(i, ~paste0(., "_otu_tax")) tmp_md_name <- purrr::map_chr(i, ~paste0(., "_md")) tmp_tax_table_fp <-paste(tmp_path, tmp_otu_name, ".txt", sep ="") tmp_map_fp <-paste(tmp_path, tmp_md_name, ".txt", sep ="") tmp_input <-load_taxa_table(tmp_tax_table_fp, tmp_map_fp) tmp_input_name <- purrr::map_chr(i, ~paste0(., "_mc"))print(tmp_input_name)assign(tmp_input_name, tmp_input)rm(list =ls(pattern ="tmp_"))}rm(list =ls(pattern ="_md"))rm(list =ls(pattern ="_otu_tax"))```## Choose Data SetAt this point in the code we need to choose a data set to use, formatted with `mctoolsr`. Remember, there are four choices:1) FULL, unfiltered data set.2) Arbitrary filtered data set.3) PERfect filtered data set.4) PIME filtered data set.This way, if we want to test other data sets we only need to change the name here.```{r}objects(pattern ="_mc")ssu18_select_mc <- ssu18_ps_pime_mc```## Normality TestsBefore proceeding, we need to test each parameter in the metadata to see which ones are and are not normally distributed. For that, we use the Shapiro-Wilk Normality Test. Here we only need one of the metadata files.```{r}#| code-fold: truetemp_md <- ssu18_select_mc$map_loadedtemp_md[,1:9] <-NULLshap_results <-NULLfor (i incolnames(temp_md)) { tmp_shap <-shapiro.test(temp_md[[i]]) tmp_p <-round(tmp_shap$p.value, digits =5) tmp_res <-eval(isTRUE(tmp_shap$p.value <0.05)) shap_results <-rbind(shap_results, data.frame(i, tmp_p, tmp_res))rm(list =ls(pattern ="tmp_"))}colnames(shap_results) <-c("parameter", "p-value", "tranform")shap_resultsdplyr::filter(shap_results, tranform =="TRUE")md_to_tranform <- shap_results$parameter[shap_results$tranform ==TRUE]rm(list =ls(pattern ="temp_md"))```<details><summary>**Show the results** of each normality test for metadata parameters</summary></br><small>`r caption_tab_ssu("ssu_normality_tests")`</small>```{r}#| echo: false#| eval: trueseq_table <- shap_resultsreactable(seq_table,defaultColDef =colDef(headerStyle =list(fontSize ="0.8em"),header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =1),align ="center", filterable =TRUE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold") ), columns =list(parameter =colDef(name ="Parameter", sticky ="left", style =list(borderRight ="1px solid #eee"),headerStyle =list(borderRight ="1px solid #eee", fontSize ="0.9em"), align ="left",minWidth =150), `p-value`=colDef(name ="p-value", style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color, fontWeight ="bold")}) ), searchable =TRUE, defaultPageSize =60, pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =FALSE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =FALSE, wrap =FALSE, showSortable =TRUE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```</details>Looks like we need to transform `r length(md_to_tranform)` metadata parameters.## Normalize ParametersHere we use the R package [`bestNormalize`](https://cran.r-project.org/web/packages/bestNormalize/vignettes/bestNormalize.html) to find and execute the best normalizing transformation. The function will test the following normalizing transformations:- `arcsinh_x` performs an arcsinh transformation.- `boxcox` Perform a Box-Cox transformation and center/scale a vector to attempt normalization. `boxcox` estimates the optimal value of lambda for the Box-Cox transformation. The function will return an error if a user attempt to transform nonpositive data.- `yeojohnson` Perform a Yeo-Johnson Transformation and center/scale a vector to attempt normalization. `yeojohnson` estimates the optimal value of lambda for the Yeo-Johnson transformation. The Yeo-Johnson is similar to the Box-Cox method, however it allows for the transformation of nonpositive data as well. - `orderNorm` The Ordered Quantile (ORQ) normalization transformation, `orderNorm()`, is a rank-based procedure by which the values of a vector are mapped to their percentile, which is then mapped to the same percentile of the normal distribution. Without the presence of ties, this essentially guarantees that the transformation leads to a uniform distribution.- `log_x` performs a simple log transformation. The parameter a is essentially estimated by the training set by default (estimated as the minimum possible to some extent epsilon), while the base must be specified beforehand. The default base of the log is 10.- `sqrt_x` performs a simple square-root transformation. The parameter a is essentially estimated by the training set by default (estimated as the minimum possible), while the base must be specified beforehand.- `exp_x` performs a simple exponential transformation.See this GitHub issue ([#5](https://github.com/petersonR/bestNormalize/issues/5)) for a description on getting reproducible results. Apparently, you can get different results because the `bestNormalize()` function uses repeated cross-validation (and doesn't automatically set the seed), so the results will be slightly different each time the function is executed.```{r}#| code-fold: trueset.seed(119)for (i in md_to_tranform) { tmp_md <- ssu18_select_mc$map_loaded tmp_best_norm <-bestNormalize(tmp_md[[i]], r =1, k =5, loo =TRUE) tmp_name <- purrr::map_chr(i, ~paste0(., "_best_norm_test"))assign(tmp_name, tmp_best_norm)print(tmp_name)rm(list =ls(pattern ="tmp_"))}```<details><summary>Show the chosen transformations</summary>```{r}#| echo: false#| eval: truefor (i in md_to_tranform) { tmp_get <-get(purrr::map_chr(i, ~paste0(., "_best_norm_test"))) tmp_print <-c("bestNormalize Chosen transformation of", i)cat("\n")cat("##", tmp_print, "##", "\n")print(tmp_get$chosen_transform)cat("_____________________________________")cat("\n")rm(list =ls(pattern ="tmp_"))}```</details><details><summary>Show the complete `bestNormalize` results</summary>```{r}#| echo: false#| eval: truefor (i in md_to_tranform) { tmp_get <-get(purrr::map_chr(i, ~paste0(., "_best_norm_test"))) tmp_print <-c("Results of bestNormalize for", i)cat("\n")cat("##", tmp_print, "##", "\n")print(tmp_get)cat("_____________________________________")cat("\n")rm(list =ls(pattern ="tmp_"))}```</details>Great, now we can add the normalized transformed data back to our `mctoolsr` metadata file. ```{r}#| code-fold: truessu18_select_mc_norm <- ssu18_select_mcfor (i in md_to_tranform) { tmp_get <-get(purrr::map_chr(i, ~paste0(i, "_best_norm_test"))) tmp_new_data <- tmp_get$x.t ssu18_select_mc_norm$map_loaded[[i]] <- tmp_new_datarm(list =ls(pattern ="tmp_"))} ```And rerun the Shapiro Tests. ```{r}#| code-fold: truetemp_md_norm <- ssu18_select_mc_norm$map_loadedtemp_md_norm[,1:9] <-NULLshap_results_norm <-NULLfor (i incolnames(temp_md_norm)) { tmp_shap <-shapiro.test(temp_md_norm[[i]]) tmp_p <-round(tmp_shap$p.value, digits =5) tmp_res <-eval(isTRUE(tmp_shap$p.value <0.05)) shap_results_norm <-rbind(shap_results_norm, data.frame(i, tmp_p, tmp_res))rm(list =ls(pattern ="tmp_"))}colnames(shap_results_norm) <-c("parameter", "p-value", "tranform")shap_results_normrm(list =ls(pattern ="temp_md_norm"))```And check if there are any parameters that are still significant for the normality test.```{r}#| eval: trueshap_results$parameter[shap_results_norm$tranform ==TRUE]```Ok. Looks like `bestNormalize` was unable to find a suitable transformation for `Al` and `Fe`. This is likely because there is very little variation in these metadata and/or there are too few significant digits.## Normalized MetadataFinally, here is a new summary table that includes all of the normalized data.</br>```{r}#| echo: false#| eval: truemetad_norm <- ssu18_select_mc_norm$map_loadedmetad_norm <- metad_norm %>% tibble::rownames_to_column("Sample_ID")metad_norm <- metad_norm[order(match(metad_norm$Sample_ID, metad$Sample_ID)), ]metad_norm[,2:9] <-NULLseq_table <- metad_normwrite.table(metad_norm, "files/metadata/tables/ssu18_normalized_metadata.txt", row.names =FALSE, sep ="\t", quote =FALSE)```<small>`r caption_tab_ssu("ssu_normalized_metadata")`</small>```{r}#| echo: false#| eval: trueseq_table <- seq_table %>% dplyr::mutate(across(.cols =c(3:5, 7:9, 14:15,19:20, 24,26,30,34:38,40, 42:43, 49, 52:53,55,57,60:62), round, digits =3)) reactable(seq_table,defaultColDef =colDef(headerStyle =list(fontSize ="0.8em"),header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =1),align ="center", filterable =TRUE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold") ), columns =list(Sample_ID =colDef(name ="Sample ID", sticky ="left", style =list(borderRight ="1px solid #eee"),headerStyle =list(borderRight ="1px solid #eee", fontSize ="0.9em"), align ="left",minWidth =150), raw_reads =colDef(footer =function(values) sprintf("%.0f", sum(values))) ), searchable =TRUE, defaultPageSize =5, pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =FALSE, wrap =FALSE, showSortable =TRUE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```{{< downloadthis files/metadata/tables/ssu18_normalized_metadata.txt dname="ssu18_normalized_metadata" label="Download table as text file" icon="table" type="primary" >}}```{r}#| echo: false#| eval: false#| comment: "Code to run individual transformations for each metadata and generate histograms" ##for (i in md_to_tranform) {## tmp_data <- ssu18_select_mc$map_loaded## tmp_no_transform_obj <- no_transform(tmp_data[[i]])## tmp_arcsinh_obj <- arcsinh_x(tmp_data[[i]])## #tmp_boxcox_obj <- boxcox(tmp_data[[i]])## tmp_yeojohnson_obj <- yeojohnson(tmp_data[[i]])## tmp_orderNorm_obj <- orderNorm(tmp_data[[i]])## tmp_log_obj <- log_x(tmp_data[[i]])## tmp_sqrt_x_obj <- sqrt_x(tmp_data[[i]])## tmp_exp_x_obj <- exp_x(tmp_data[[i]])## ## tmp_plot_no_transform <- hist(tmp_no_transform_obj$x.t, main = i, breaks = 6, xlab = "no_transform")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_no_transform"))## assign(tmp_plot_name, tmp_plot_no_transform)## print(tmp_plot_name)## ## tmp_plot_arcsinh <- hist(tmp_arcsinh_obj$x.t, main = i, breaks = 6, xlab = "arcsinh")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_arcsinh"))## assign(tmp_plot_name, tmp_plot_arcsinh)## print(tmp_plot_name)## # cannot run because of Al and Fe## #tmp_plot_boxcox <- hist(tmp_boxcox_obj$x.t, main = i, breaks = 6, xlab = "boxcox")## #tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_boxcox"))## #assign(tmp_plot_name, tmp_plot_boxcox)## #print(tmp_plot_name)## ## tmp_plot_yeojohnson <- hist(tmp_yeojohnson_obj$x.t, main = i, breaks = 6, xlab = "yeojohnson")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_yeojohnson"))## assign(tmp_plot_name, tmp_plot_yeojohnson)## print(tmp_plot_name)## ## tmp_plot_orderNorm <- hist(tmp_orderNorm_obj$x.t, main = i, breaks = 6, xlab = "orderNorm")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_orderNorm"))## assign(tmp_plot_name, tmp_plot_orderNorm)## print(tmp_plot_name)## ## tmp_plot_log <- hist(tmp_log_obj$x.t, main = i, breaks = 6, xlab = "log")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_log"))## assign(tmp_plot_name, tmp_plot_log)## print(tmp_plot_name)## ## tmp_plot_sqrt <- hist(tmp_sqrt_x_obj$x.t, main = i, breaks = 6, xlab = "sqrt")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_sqrt"))## assign(tmp_plot_name, tmp_plot_sqrt)## print(tmp_plot_name)## ## tmp_plot_exp <- hist(tmp_exp_x_obj$x.t, main = i, breaks = 6, xlab = "exp")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_exp"))## assign(tmp_plot_name, tmp_plot_exp)## print(tmp_plot_name)#### rm(list = ls(pattern = "tmp_"))##}```## Autocorrelation TestsNext, we test the metadata for autocorrelations. Do we do this on the original data or the transformed data? No idea, so let's do both.### Split MetadataWe need to split the data into different groups.A) Environmental and edaphic propertiesB) Microbial functional responsesC) Temperature adaptation propertiesWe first create lists of metadata parameters. ```{r}#| code-fold: truediv <-c("PLOT", "TREAT", "TREAT_T", "PAIR", "Observed", "Shannon_exp", "InvSimpson", "ATAP")edaphic <-c("AST", "H2O", "N", "P", "Al", "Ca", "Fe", "K", "Mg", "Mn", "Na", "TEB", "ECEC", "pH", "NH4", "NO3", "PO4", "DOC", "DON", "DOCN")soil_funct <-c("micC", "micN", "micP", "micCN", "micCP", "micNP", "AG_ase", "BG_ase", "BP_ase", "CE_ase", "P_ase", "N_ase", "S_ase", "XY_ase", "LP_ase", "PX_ase", "CO2", "enzCN", "enzCP", "enzNP")temp_adapt <-c("AG_Q10", "BG_Q10", "BP_Q10", "CE_Q10", "P_Q10", "N_Q10", "S_Q10", "XY_Q10", "LP_Q10", "PX_Q10", "CUEcn", "CUEcp", "NUE","PUE", "Tmin", "SI")md_groups <-c("edaphic", "soil_funct", "temp_adapt")# NoT uSed: minPO4, minNH4, minNO3, minTIN```And then use the lists to split the data sets by metadata group. Here, we do this for the original metadata and the metadata after normalization.```{r}#| code-fold: trueselect_md <-c("ssu18_select_mc", "ssu18_select_mc_norm")for (i in select_md) {#tmp_get <- get(purrr::map_chr(i, ~ paste0(i, "_mc"))) tmp_get <-get(i) tmp_md_all <- tmp_get$map_loaded tmp_div <- tmp_md_all %>% dplyr::select(all_of(div)) tmp_div <- tmp_div %>% tibble::rownames_to_column("SampleID")## edaphic tmp_sub_edaphic <- tmp_md_all %>% dplyr::select(all_of(edaphic)) tmp_sub_edaphic <- tmp_sub_edaphic %>% tibble::rownames_to_column("SampleID") tmp_edaphic <- dplyr::left_join(tmp_div, tmp_sub_edaphic, by ="SampleID") tmp_edaphic <- tmp_edaphic %>% tibble::column_to_rownames("SampleID")## soil_funct tmp_sub_soil_funct <- tmp_md_all %>% dplyr::select(all_of(soil_funct)) tmp_sub_soil_funct <- tmp_sub_soil_funct %>% tibble::rownames_to_column("SampleID") tmp_soil_funct <- dplyr::left_join(tmp_div, tmp_sub_soil_funct, by ="SampleID") tmp_soil_funct <- tmp_soil_funct %>% tibble::column_to_rownames("SampleID") ## temp_adapt tmp_sub_temp_adapt <- tmp_md_all %>% dplyr::select(all_of(temp_adapt)) tmp_sub_temp_adapt <- tmp_sub_temp_adapt %>% tibble::rownames_to_column("SampleID") tmp_temp_adapt <- dplyr::left_join(tmp_div, tmp_sub_temp_adapt, by ="SampleID") tmp_temp_adapt <- tmp_temp_adapt %>% tibble::column_to_rownames("SampleID") ## combine tmp_list <-list(data_loaded = ssu18_select_mc$data_loaded, map_loaded = ssu18_select_mc$map_loaded, taxonomy_loaded = ssu18_select_mc$taxonomy_loaded,edaphic = tmp_edaphic, soil_funct = tmp_soil_funct, temp_adapt = tmp_temp_adapt) tmp_name <- purrr::map_chr(i, ~paste0(., "_split"))print(tmp_name)assign(tmp_name, tmp_list)rm(list =ls(pattern ="tmp_"))}```### Generate Autocorrelation PlotsA little housekeeping to get rid of parameters we don't need (e.g., plot number, pair, etc.).```{r}#| code-fold: trueedaphic_cor <- ssu18_select_mc_split$edaphicedaphic_cor[,1:8] <-NULLedaphic_norm_cor <- ssu18_select_mc_norm_split$edaphicedaphic_norm_cor[,1:8] <-NULLsoil_funct_cor <- ssu18_select_mc_split$soil_functsoil_funct_cor[,1:8] <-NULLsoil_funct_norm_cor <- ssu18_select_mc_norm_split$soil_functsoil_funct_norm_cor[,1:8] <-NULLtemp_adapt_cor <- ssu18_select_mc_split$temp_adapttemp_adapt_cor[,1:8] <-NULLtemp_adapt_norm_cor <- ssu18_select_mc_norm_split$temp_adapttemp_adapt_norm_cor[,1:8] <-NULL```And finally the code to create the plots. ```{r}#| code-fold: truefor (i inobjects(pattern ="_cor$")) { tmp_get <-get(i) tmp_cormat <-round(cor(tmp_get), 2) tmp_melted_cormat <- reshape2::melt(tmp_cormat) tmp_get_lower_tri <-function(tmp_cormat){ tmp_cormat[upper.tri(tmp_cormat)] <-NAreturn(tmp_cormat) }# Get upper triangle of the correlation matrix tmp_get_upper_tri <-function(tmp_cormat){ tmp_cormat[lower.tri(tmp_cormat)] <-NAreturn(tmp_cormat) } tmp_upper_tri <-tmp_get_upper_tri(tmp_cormat) tmp_melted_cormat <- reshape2::melt(tmp_upper_tri, na.rm =TRUE)ggplot(data = tmp_melted_cormat, aes(x = Var1, y = Var2, fill = value)) +geom_tile() tmp_ggheatmap <-ggplot(data = tmp_melted_cormat, aes(Var2, Var1, fill = value)) +geom_tile(color ="white") +scale_fill_gradient2(low ="blue", high ="red", mid ="white", midpoint =0, limit =c(-1,1), space ="Lab", name="Pearson\nCorrelation") +theme_minimal() +theme(axis.text.x =element_text(angle =45, vjust =1, size =7, hjust =1),axis.text.y =element_text(vjust =1, size =7, hjust =1)) +coord_fixed() +geom_text(aes(Var2, Var1, label = value), color ="black", size =1.75) +theme(axis.title.x =element_blank(),axis.title.y =element_blank(),panel.grid.major =element_blank(),panel.border =element_blank(),panel.background =element_blank(),axis.ticks =element_blank(),legend.justification =c(1, 0),legend.position =c(0.6, 0.7),legend.direction ="horizontal") +guides(fill =guide_colorbar(barwidth =7, barheight =1,title.position ="top", title.hjust =0.5)) tmp_name <- purrr::map_chr(i, ~paste0(., "_ggheatmap"))assign(tmp_name, tmp_ggheatmap)print(tmp_name)rm(list =ls(pattern ="tmp_"))} objects(pattern ="_ggheatmap")```## Autocorrelation Plots<br/>::: {.column-body}::: {.panel-tabset}#### Edaphic properties```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5edaphic_cor_ggheatmap```<small>`r caption_fig_ssu("ssu_autocor_edaph_plots")`</small>#### Edaphic properties (normalized)```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5edaphic_norm_cor_ggheatmap```<small>`r caption_fig_ssu("ssu_autocor_edaph_norm_plots")`</small>:::::: {.panel-tabset}#### Functional responses```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5soil_funct_cor_ggheatmap```<small>`r caption_fig_ssu("ssu_autocor_funct_plots")`</small>#### Functional responses (normalized)```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5soil_funct_norm_cor_ggheatmap```<small>`r caption_fig_ssu("ssu_autocor_funct_norm_plots")`</small>:::::: {.panel-tabset}#### Temperature adaptation ```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5temp_adapt_cor_ggheatmap```<small>`r caption_fig_ssu("ssu_autocor_temp_plots")`</small>#### Temperature adaptation (normalized)```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5temp_adapt_norm_cor_ggheatmap```<small>`r caption_fig_ssu("ssu_autocor_temp_norm_plots")`</small>::::::```{r}#| echo: falseauto_cor_figs <-ggarrange( edaphic_cor_ggheatmap, edaphic_norm_cor_ggheatmap, soil_funct_cor_ggheatmap, soil_funct_norm_cor_ggheatmap, temp_adapt_cor_ggheatmap, temp_adapt_norm_cor_ggheatmap,ncol =2, nrow =3, common.legend =FALSE)auto_cor_figsdev.off()png("files/metadata/figures/ssu18_auto_cor_figs.png",height =32, width =24, units ='cm', res =600, bg ="white")auto_cor_figsdev.off()pdf("files/metadata/figures/ssu18_auto_cor_figs.pdf",height =10, width =12)auto_cor_figsdev.off()```Now we can remove parameters based on the autocorrelation analysis:A) Environmental and edaphic properties: TEB, DON, Na, Al, Ca.B) Microbial functional responses: micN, micNP, enzCN, enzCP, BP~ase~, CE~ase~, LP~ase~, N~ase~, P~ase~.C) Temperature adaptation properties: NUE, PUE, SI.```{r}#| echo: false#| eval: false# FOR TESTINGobjects(pattern ="_cor_ggheatmap")edaphic_norm_cor_ggheatmapsoil_funct_norm_cor_ggheatmaptemp_adapt_norm_cor_ggheatmapdim(edaphic_norm_cor)dim(soil_funct_norm_cor)dim(temp_adapt_norm_cor)tmp_res <- temp_adapt_norm_cor_ggheatmap$datatmp_res %>%filter(value >0.7| value <-0.7) %>%filter_("Var1 != Var2")#tmp_res %>% filter(Var1 == "DOCN" | Var2 == "DOCN") %>% filter_("Var1 != Var2")write.table(tmp_res %>%filter(value >0.7| value <-0.7) %>%filter_("Var1 != Var2"), "soil_funct_norm7.txt", sep ="\t", quote =FALSE, row.names =FALSE)``````{r}edaphic_remove <-c("TEB", "DON", "Na", "Al", "Ca")soil_funct_remove <-c("micN", "micNP", "enzCN", "enzCP", "BP_ase", "CE_ase", "LP_ase", "N_ase", "P_ase")temp_adapt_remove <-c("NUE", "PUE", "SI")``````{r}#| code-fold: truetmp_df <- ssu18_select_mc_splittmp_df$edaphic <- tmp_df$edaphic[, !names(tmp_df$edaphic) %in% edaphic_remove]tmp_df$soil_funct <- tmp_df$soil_funct[, !names(tmp_df$soil_funct) %in% soil_funct_remove]tmp_df$temp_adapt <- tmp_df$temp_adapt[, !names(tmp_df$temp_adapt) %in% temp_adapt_remove]ssu18_select_mc_split_no_ac <- tmp_dfrm(list =ls(pattern ="tmp_"))tmp_df <- ssu18_select_mc_norm_splittmp_df$edaphic <- tmp_df$edaphic[, !names(tmp_df$edaphic) %in% edaphic_remove]tmp_df$soil_funct <- tmp_df$soil_funct[, !names(tmp_df$soil_funct) %in% soil_funct_remove]tmp_df$temp_adapt <- tmp_df$temp_adapt[, !names(tmp_df$temp_adapt) %in% temp_adapt_remove]ssu18_select_mc_norm_split_no_ac <- tmp_dfrm(list =ls(pattern ="tmp_"))```## Dissimilarity Correlation TestsLet's see if any on the metadata groups are significantly correlated with the community data. Basically, we create distance matrices for the community data and each metadata group and then run Mantel tests for all comparisons. For the community data we calculate Bray-Curtis distances for the community data and Euclidean distances for the metadata. We use the function `mantel.test` from the `ape` package and `mantel` from the `vegan` package for the analyses. In summary, we test both `mantel.test` and `mantel` on Bray-Curtis distance community distances against Euclidean distances for each metadata group (`edaphic`, `soil_funct`, `temp_adapt`) **a**) before normalizing and before removing autocorrelated parameters, **b**) before normalizing and after removing autocorrelated parameters, **c**) after normalizing and before removing autocorrelated parameters, and **d**) after normalizing and after removing autocorrelated parameters.```{r}#| code-fold: trueman_df <-c("ssu18_select_mc_split", "ssu18_select_mc_split_no_ac", "ssu18_select_mc_norm_split", "ssu18_select_mc_norm_split_no_ac")for (i in man_df) { tmp_get <-get(i) tmp_dm_otu <-as.matrix(vegdist(t(tmp_get$data_loaded), method ="bray", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE))# EDAPHIC tmp_dm_md_edaphic <-as.matrix(vegdist(tmp_get$edaphic[, 8:ncol(tmp_get$edaphic)], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man1_edaphic <-mantel.test(tmp_dm_otu, tmp_dm_md_edaphic, nperm =999, graph =FALSE, alternative ="two.sided") tmp_man2_edaphic <-mantel(tmp_dm_otu, tmp_dm_md_edaphic, permutations =999)# SOIL FUNCT tmp_dm_md_soil_funct <-as.matrix(vegdist(tmp_get$soil_funct[, 8:ncol(tmp_get$soil_funct)], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man1_soil_funct <-mantel.test(tmp_dm_otu, tmp_dm_md_soil_funct, nperm =999, graph =FALSE, alternative ="two.sided") tmp_man2_soil_funct <-mantel(tmp_dm_otu, tmp_dm_md_soil_funct, permutations =999)# TEMP ADAPT tmp_dm_md_temp_adapt <-as.matrix(vegdist(tmp_get$temp_adapt[, 8:ncol(tmp_get$temp_adapt)], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man1_temp_adapt <-mantel.test(tmp_dm_otu, tmp_dm_md_temp_adapt, nperm =999, graph =FALSE, alternative ="two.sided") tmp_man2_temp_adapt <-mantel(tmp_dm_otu, tmp_dm_md_temp_adapt, permutations =999) tmp_name <- purrr::map_chr(i, ~paste0(., "_mantel_tests")) tmp_df <-list(edaphic_ape_man = tmp_man1_edaphic, edaphic_vegan_man = tmp_man2_edaphic,soil_funct_ape_man = tmp_man1_soil_funct, soil_funct_vegan_man = tmp_man2_soil_funct,temp_adapt_ape_man = tmp_man1_temp_adapt, temp_adapt_vegan_man = tmp_man2_temp_adapt)assign(tmp_name, tmp_df)print(tmp_name)rm(list =ls(pattern ="tmp_"))}``````{r}#| echo: falsetmp_objects <-c("ssu18_select_mc_split_mantel_tests", "ssu18_select_mc_split_no_ac_mantel_tests","ssu18_select_mc_norm_split_mantel_tests","ssu18_select_mc_norm_split_no_ac_mantel_tests")tmp_norm <-data.frame(c("no", "no", "yes", "yes"))tmp_ac <-data.frame(c("no", "yes", "no", "yes"))ssu18_ps_pime_mantel_summary <- dplyr::bind_cols(tmp_norm, tmp_ac) %>% dplyr::rename("normalized?"=1) %>% dplyr::rename("AC removed?"=2)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$edaphic_ape_man$p,get(tmp_objects[2])$edaphic_ape_man$p,get(tmp_objects[3])$edaphic_ape_man$p,get(tmp_objects[4])$edaphic_ape_man$p))ssu18_ps_pime_mantel_summary <- dplyr::bind_cols(ssu18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("edaphic_ape"=3)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$edaphic_vegan_man$signif,get(tmp_objects[2])$edaphic_vegan_man$signif,get(tmp_objects[3])$edaphic_vegan_man$signif,get(tmp_objects[4])$edaphic_vegan_man$signif))ssu18_ps_pime_mantel_summary <- dplyr::bind_cols(ssu18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("edaphic_vegan"=4)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$soil_funct_ape_man$p,get(tmp_objects[2])$soil_funct_ape_man$p,get(tmp_objects[3])$soil_funct_ape_man$p,get(tmp_objects[4])$soil_funct_ape_man$p))ssu18_ps_pime_mantel_summary <- dplyr::bind_cols(ssu18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("soil_funct_ape"=5)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$soil_funct_vegan_man$signif,get(tmp_objects[2])$soil_funct_vegan_man$signif,get(tmp_objects[3])$soil_funct_vegan_man$signif,get(tmp_objects[4])$soil_funct_vegan_man$signif))ssu18_ps_pime_mantel_summary <- dplyr::bind_cols(ssu18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("soil_funct_vegan"=6)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$temp_adapt_ape_man$p,get(tmp_objects[2])$temp_adapt_ape_man$p,get(tmp_objects[3])$temp_adapt_ape_man$p,get(tmp_objects[4])$temp_adapt_ape_man$p))ssu18_ps_pime_mantel_summary <- dplyr::bind_cols(ssu18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("temp_adapt_ape"=7)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$temp_adapt_vegan_man$signif,get(tmp_objects[2])$temp_adapt_vegan_man$signif,get(tmp_objects[3])$temp_adapt_vegan_man$signif,get(tmp_objects[4])$temp_adapt_vegan_man$signif))ssu18_ps_pime_mantel_summary <- dplyr::bind_cols(ssu18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("temp_adapt_vegan"=8)rm(list =ls(pattern ="tmp_"))```### Dissimilarity Correlation Results<br/><small>`r caption_tab_ssu("ssu_mantel_summ")`</small>```{r}#| echo: false#| eval: trueseq_table <- ssu18_ps_pime_mantel_summaryreactable(seq_table,defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =3),align ="center", filterable =FALSE, sortable =FALSE, resizable =TRUE, minWidth =50,style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color)} ), columns =list(`normalized?`=colDef(name ="normalized?", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee"), align ="left",minWidth =70 ),`AC removed?`=colDef(name ="AC removed?", align ="left",minWidth =70,headerStyle =list(borderRight ="5px solid #eee"), style =list(borderRight ="5px solid #eee") ),edaphic_ape =colDef(name ="ape", ),edaphic_vegan =colDef(name ="vegan" ),soil_funct_ape =colDef(name ="ape" ),soil_funct_vegan =colDef(name ="vegan" ),temp_adapt_ape =colDef(name ="ape" ),temp_adapt_vegan =colDef(name ="vegan" ) ), columnGroups =list(colGroup(name ="Environmental & edaphic", columns =c("edaphic_ape", "edaphic_vegan"), headerStyle =list(fontSize ="1.2em" ), ),colGroup(name ="Functional responses", columns =c("soil_funct_ape", "soil_funct_vegan"), headerStyle =list(fontSize ="1.2em" ) ),colGroup(name ="Temperature adaptation", columns =c("temp_adapt_ape", "temp_adapt_vegan"), headerStyle =list(fontSize ="1.2em") ) ),searchable =FALSE, defaultPageSize =15, showPagination =FALSE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =FALSE, showSortable =FALSE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```Moving on.## Best Subset of VariablesNow we want to know which of the metadata parameters are the most strongly correlated with the community data. For this we use the `bioenv` function from the `vegan` package. `bioenv`---*Best Subset of Environmental Variables with Maximum (Rank) Correlation with Community Dissimilarities*---finds the best subset of environmental variables, so that the Euclidean distances of scaled environmental variables have the maximum (rank) correlation with community dissimilarities. Next we will use the metadata set where autocorrelated parameters were removed and the remainder of the parameters were normalized (where applicable based on the Shapiro tests). We run `bioenv` against the three groups of metadata parameters. We then run `bioenv` again, but this time against the individual parameters identified as significantly correlated.### Edaphic Properties```{r}#| code-fold: truetmp_comm <-data.frame(t(ssu18_select_mc_norm_split_no_ac$data_loaded))tmp_env <-data.frame(ssu18_select_mc_norm_split_no_ac$edaphic)tmp_env[,1:8] <-NULLedaphic_bioenv <-bioenv(wisconsin(tmp_comm), tmp_env, method ="spearman", index ="bray", upto =ncol(tmp_env), metric ="euclidean")bioenv_list <- edaphic_bioenv$models[[edaphic_bioenv$whichbest]]$bestbioenv_best <-bioenvdist(edaphic_bioenv, which ="best")for (i in bioenv_list) { tmp_dp <-data.frame(edaphic_bioenv$x) tmp_md <-as.matrix(vegdist(tmp_dp[[i]], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man <-mantel(bioenv_best, tmp_md, permutations =999, method ="spearman") tmp_md_name <-names(tmp_dp)[[i]] tmp_name <- purrr::map_chr(tmp_md_name, ~paste0(., "_bioenv_mantel_test"))assign(tmp_name, tmp_man)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bioenv_mantel_test")edaphic_bioenv_ind_mantel <-list(AST = AST_bioenv_mantel_test)rm(list =ls(pattern ="_bioenv_mantel_test"))``````{r}#| echo: false#| eval: trueedaphic_bioenv```<details><summary>Show the results of individual edaphic metadata Mantel tests</summary>```{r}#| echo: false#| eval: trueedaphic_bioenv_ind_mantel```</details>`bioenv` found the following edaphic properties significantly correlated with the community data: **`r row.names(summary(edaphic_bioenv_ind_mantel))`**### Soil Functional Response```{r}#| code-fold: truetmp_comm <-data.frame(t(ssu18_select_mc_norm_split_no_ac$data_loaded))tmp_env <-data.frame(ssu18_select_mc_norm_split_no_ac$soil_funct)tmp_env[,1:8] <-NULLsoil_funct_bioenv <-bioenv(wisconsin(tmp_comm), tmp_env, method ="spearman", index ="bray", upto =ncol(tmp_env), metric ="euclidean")bioenv_list <- soil_funct_bioenv$models[[soil_funct_bioenv$whichbest]]$bestbioenv_best <-bioenvdist(soil_funct_bioenv, which ="best")for (i in bioenv_list) { tmp_dp <-data.frame(soil_funct_bioenv$x) tmp_md <-as.matrix(vegdist(tmp_dp[[i]], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man <-mantel(bioenv_best, tmp_md, permutations =999, method ="spearman") tmp_md_name <-names(tmp_dp)[[i]] tmp_name <- purrr::map_chr(tmp_md_name, ~paste0(., "_bioenv_mantel_test"))assign(tmp_name, tmp_man)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bioenv_mantel_test")soil_funct_bioenv_ind_mantel <-list(AG_ase = AG_ase_bioenv_mantel_test, enzNP = enzNP_bioenv_mantel_test, S_ase = S_ase_bioenv_mantel_test, PX_ase = PX_ase_bioenv_mantel_test,XY_ase = XY_ase_bioenv_mantel_test)rm(list =ls(pattern ="_bioenv_mantel_test"))``````{r}#| echo: false#| eval: truesoil_funct_bioenv```<details><summary>Show the results of individual functional response metadata Mantel tests</summary>```{r}#| echo: false#| eval: truesoil_funct_bioenv_ind_mantel```</details>`bioenv` found the following soil functions significantly correlated with the community data: **`r row.names(summary(soil_funct_bioenv_ind_mantel))`**### Temperature Adaptation```{r}#| code-fold: truetmp_comm <-data.frame(t(ssu18_select_mc_norm_split_no_ac$data_loaded))tmp_env <-data.frame(ssu18_select_mc_norm_split_no_ac$temp_adapt)tmp_env[,1:8] <-NULLtemp_adapt_bioenv <-bioenv(wisconsin(tmp_comm), tmp_env, method ="spearman", index ="bray", upto =ncol(tmp_env), metric ="euclidean")bioenv_list <- temp_adapt_bioenv$models[[temp_adapt_bioenv$whichbest]]$bestbioenv_best <-bioenvdist(temp_adapt_bioenv, which ="best")for (i in bioenv_list) { tmp_dp <-data.frame(temp_adapt_bioenv$x) tmp_md <-as.matrix(vegdist(tmp_dp[[i]], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man <-mantel(bioenv_best, tmp_md, permutations =999, method ="spearman") tmp_md_name <-names(tmp_dp)[[i]] tmp_name <- purrr::map_chr(tmp_md_name, ~paste0(., "_bioenv_mantel_test"))assign(tmp_name, tmp_man)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bioenv_mantel_test")temp_adapt_bioenv_ind_mantel <-list(CUEcp = CUEcp_bioenv_mantel_test, LP_Q10 = LP_Q10_bioenv_mantel_test, P_Q10 = P_Q10_bioenv_mantel_test, S_Q10 = S_Q10_bioenv_mantel_test, Tmin = Tmin_bioenv_mantel_test)rm(list =ls(pattern ="_bioenv_mantel_test"))``````{r}#| echo: false#| eval: truetemp_adapt_bioenv```<details><summary>Show the results of individual temperature adaptation metadata Mantel tests</summary>```{r}#| echo: false#| eval: truetemp_adapt_bioenv_ind_mantel```</details>`bioenv` found the following temperature adaptations significantly correlated with the community data: **`r row.names(summary(temp_adapt_bioenv_ind_mantel))`**## Distance-based RedundancyNow we turn our attention to distance-based redundancy analysis (dbRDA), an ordination method similar to Redundancy Analysis (rda) but it allows non-Euclidean dissimilarity indices, such as Manhattan or Bray–Curtis distance. For this, we use `capscale` from the `vegan` package. `capscale` is a constrained versions of metric scaling (principal coordinates analysis), which are based on the Euclidean distance but can be used, and are more useful, with other dissimilarity measures. The functions can also perform unconstrained principal coordinates analysis, optionally using extended dissimilarities.For each of the three metadata subsets, we perform the following steps:1) Run `rankindex` to compare metadata and community dissimilarity indices for gradient detection. This will help us select the best dissimilarity metric to use.2) Run `capscale` for distance-based redundancy analysis.3) Run `envfit` to fit environmental parameters onto the ordination. This function basically calculates correlation scores between the metadata parameters and the ordination axes. 4) Select metadata parameters significant for `bioenv` (see above) and/or `envfit` analyses.5) Run `envfit` on ASVs.6) Plot the ordination and vector overlays. ### Edaphic Properties```{r}#| code-fold: truetmp_md <- ssu18_select_mc_norm_split_no_ac$edaphictmp_md$TREAT_T <-as.character(tmp_md$TREAT_T)tmp_comm <-data.frame(t(ssu18_select_mc_norm_split_no_ac$data_loaded))edaphic_rank <-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow","bra", "kul"), stepacross =FALSE, method ="spearman")``````{r}#| echo: false#| eval: trueedaphic_rank```Let's run `capscale` using Bray-Curtis. Note, we have `r ncol(ssu18_select_mc_norm_split_no_ac$edaphic) - 8` metadata parameters in this group but, for some reason, `capscale` only works with 13 parameters. This may have to do with degrees of freedom? * Starting properties: AST, H2O, N, P, Al, Ca, Fe, K, Mg, Mn, Na, TEB, ECEC, pH, NH4, NO3, PO4, DOC, DON, DOCN* Autocorrelated removed: TEB, DON, Na, Al, Ca* Remove for capscale: Mg, Mn```{r}#| echo: false# All edaphic: AST, H2O, N, P, Al, Ca, Fe, K, Mg, Mn, Na, TEB, ECEC, pH, NH4, NO3, PO4, DOC, DON, DOCN# REMOVED edaphic: TEB, DON, Na, Al, Ca# Remaining edaphic: AST, H2O, N, P, Fe, K, Mg, Mn, ECEC, pH, NH4, NO3, PO4, DOC, DOCN# 15 total, only works with 13# Removed Mg + Mn# TEST with , metaMDSdist = TRUE AND/OR , metaMDS = TRUE``````{r}edaphic_cap <-capscale(tmp_comm ~ AST + H2O + N + P + Fe + K + ECEC + pH + NH4 + NO3 + PO4 + DOC + DOCN, tmp_md, dist ="bray")colnames(tmp_md)``````{r}#| echo: false#| eval: trueedaphic_cap```Now we can look at the variance against each principal component. ```{r}#| echo: false#| eval: truestats::screeplot(edaphic_cap)```And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The `ggplot` function `autoplot` stores these data in a more accessible way than the raw results from `capscale````{r}#| eval: truelibrary(ggvegan)base::plot(edaphic_cap) tmp_auto_plt <- ggplot2::autoplot(edaphic_cap, arrows =TRUE)tmp_auto_plt``````{r}#| echo: falseanova(edaphic_cap) # overall test of the significant of the analysisanova(edaphic_cap, by ="axis", perm.max =500) # test axes for significanceanova(edaphic_cap, by ="terms", permu =500) # test for sign. environ. variables```Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function `scores` which gets species or site scores from the ordination.```{r}#| code-fold: truetmp_samp_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="sites")tmp_samp_scores[,1] <-NULLtmp_samp_scores <- tmp_samp_scores %>% dplyr::rename(SampleID = Label)tmp_md_sub <- tmp_md[, 1:4]tmp_md_sub <- tmp_md_sub %>% tibble::rownames_to_column("SampleID")edaphic_plot_data <- dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")```Now we have a new data frame that contains sample details and capscale values. ```{r}#| echo: false#| eval: trueedaphic_plot_data```We can then do the same with the metadata vectors. Here though we only need the scores and parameter name. ```{r}#| code-fold: trueedaphic_md_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="biplot")edaphic_md_scores[,1] <-NULLedaphic_md_scores <- edaphic_md_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")``````{r}#| echo: false#| eval: trueedaphic_md_scores```Let's run some quick correlations of metadata with ordination axes to see which parameters are significant. For this we use the vegan function `envfit`.```{r}#| code-fold: truetmp_samp_scores_sub <- edaphic_plot_data[, 6:7]tmp_samp_scores_sub <-as.matrix(tmp_samp_scores_sub)tmp_param_list <- edaphic_md_scores$parameterstmp_md_sub <-subset(tmp_md, select = tmp_param_list)envfit_edaphic_md <-envfit(tmp_samp_scores_sub, tmp_md_sub,perm =1000, choices =c(1, 2))``````{r}#| echo: false#| eval: trueenvfit_edaphic_md``````{r}#| code-fold: trueedaphic_md_signif_hits <- base::subset(envfit_edaphic_md$vectors$pvals, c(envfit_edaphic_md$vectors$pvals <0.05& envfit_edaphic_md$vectors$r >0.4))edaphic_md_signif_hits <-data.frame(edaphic_md_signif_hits)edaphic_md_signif_hits <-rownames(edaphic_md_signif_hits)edaphic_md_signif <- edaphic_md_scores[edaphic_md_scores$parameters %in% edaphic_md_signif_hits,]edaphic_md_signif$parameters````envfit` found that `r edaphic_md_signif$parameters` were significantly correlated. Now let's see if the same parameters are significant for the `envfit` and `bioenv` analyses.```{r}#| echo: false#| eval: true#| results: hold#| comment: ''print("Significant parameters from bioenv analysis.")row.names(summary(edaphic_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")edaphic_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(edaphic_bioenv_ind_mantel)), edaphic_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(edaphic_md_signif$parameters, row.names(summary(edaphic_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")edaphic_sig_diff <- base::union(edaphic_md_signif$parameters, row.names(summary(edaphic_bioenv_ind_mantel)))edaphic_sig_diff``````{r}#| code-fold: truenew_edaphic_md_signif_hits <- edaphic_sig_diff#new_edaphic_md_signif_hits <- append(edaphic_md_signif_hits, edaphic_sig_diff)edaphic_md_signif_all <- edaphic_md_scores[edaphic_md_scores$parameters %in% new_edaphic_md_signif_hits,]```Check. Next, we run `envfit` for the ASVs.```{r}#| code-fold: trueenvfit_edaphic_asv <-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10],perm =1000, choices =c(1, 2))edaphic_asv_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="species")edaphic_asv_scores <- edaphic_asv_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")edaphic_asv_scores[,1] <-NULL``````{r}#| echo: false#| eval: trueenvfit_edaphic_asv``````{r}#| code-fold: trueedaphic_asv_signif_hits <- base::subset(envfit_edaphic_asv$vectors$pvals, c(envfit_edaphic_asv$vectors$pvals <0.05& envfit_edaphic_asv$vectors$r >0.5))edaphic_asv_signif_hits <-data.frame(edaphic_asv_signif_hits)edaphic_asv_signif_hits <-rownames(edaphic_asv_signif_hits)edaphic_asv_signif <- edaphic_asv_scores[edaphic_asv_scores$parameters %in% edaphic_asv_signif_hits,]``````{r}#| echo: false#| eval: trueedaphic_asv_signif``````{r}#| code-fold: trueedaphic_md_signif_all$variable_type <-"metadata"edaphic_asv_signif$variable_type <-"ASV"edaphic_bioplot_data <-rbind(edaphic_md_signif_all, edaphic_asv_signif)```The last thing to do is categorize parameters scores and ASV scores into different variable types for plotting.```{r}#| code-fold: trueedaphic_bioplot_data_md <-subset(edaphic_bioplot_data, edaphic_bioplot_data$variable_type =="metadata")edaphic_bioplot_data_asv <-subset(edaphic_bioplot_data, edaphic_bioplot_data$variable_type =="ASV")```<details><summary>Show the code for the plot</summary>```{r}#| echo: falseedaphic_cap_vals <-data.frame(edaphic_cap$CCA$eig[1:2])edaphic_cap1 <-signif((edaphic_cap_vals[1,] *100), digits=3)edaphic_cap2 <-signif((edaphic_cap_vals[2,] *100), digits=3)cpa1_lab <-paste("CAP1", " (", edaphic_cap1, "%)", sep ="")cpa2_lab <-paste("CAP2", " (", edaphic_cap2, "%)", sep ="")swel_col <-c("#2271B2", "#71B222", "#B22271")edaphic_plot <-ggplot(edaphic_plot_data) +geom_point(mapping =aes(x = CAP1, y = CAP2, shape = TREAT,colour = TREAT_T), size =4) +scale_colour_manual(values = swel_col) +# geom_text(data = edaphic_plot_data, aes(x = CAP1, y = CAP2, #UNCOMMENT to add sample labels# label = SampleID), size = 3) + geom_segment(aes(x =0, y =0, xend = CAP1, yend = CAP2),data = edaphic_bioplot_data_md, linetype ="solid",arrow =arrow(length =unit(0.3, "cm")), size =0.4, color ="#191919", inherit.aes =FALSE) +geom_text(data = edaphic_bioplot_data_md, aes(x = CAP1, y = CAP2, label = parameters), size =3, nudge_x =0.1, nudge_y =0.05) +### This code adds ASV vectors, Uncomment to add#geom_segment(aes(x = 0, y = 0, xend = CAP1, yend = CAP2),# data = edaphic_bioplot_data_asv, linetype = "solid",# arrow = arrow(length = unit(0.3, "cm")), size = 0.2, # color = "#676767") +#geom_text(data = edaphic_bioplot_data_asv, # aes(x = CAP1, y = CAP2, label = parameters), size = 2.5,# nudge_x = 0.1, nudge_y = 0.05) +theme_classic(base_size =12) +labs(title ="Capscale Analysis",subtitle ="Edaphic properties", x = cpa1_lab, y = cpa2_lab)edaphic_plot <- edaphic_plot +coord_fixed() +theme(aspect.ratio=1)edaphic_plotpng("files/metadata/figures/ssu18_edaphic_capscale.png",height =16, width =20, units ='cm', res =600, bg ="white")edaphic_plotinvisible(dev.off())pdf("files/metadata/figures/ssu18_edaphic_capscale.pdf",height =5, width =6)edaphic_plotdev.off()```</details>```{r}#| echo: falserm(list =ls(pattern ="tmp_"))```### Soil Functional Response```{r}#| code-fold: truetmp_md <- ssu18_select_mc_norm_split_no_ac$soil_functtmp_md$TREAT_T <-as.character(tmp_md$TREAT_T)tmp_comm <-data.frame(t(ssu18_select_mc_norm_split_no_ac$data_loaded))soil_funct_rank <-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow","bra", "kul"), stepacross =FALSE, method ="spearman")``````{r}#| echo: false#| eval: truesoil_funct_rank```Let's run `capscale` using Bray-Curtis. Note, we have `r ncol(ssu18_select_mc_norm_split_no_ac$soil_funct) - 8` metadata parameters in this group but, for some reason, `capscale` only works with 13 parameters. This may have to do with degrees of freedom? * Starting properties: micC, micN, micP, micCN, micCP, micNP, AG_ase, BG_ase, BP_ase, CE_ase, P_ase, N_ase, S_ase, XY_ase, LP_ase, PX_ase, CO2, enzCN, enzCP, enzNP* Autocorrelated removed: micN, micNP, enzCN, enzCP, BP_ase, CE_ase, LP_ase, N_ase, P_ase* Remove for capscale: NONE```{r}#| echo: false# All soil prop: micC, micN, micP, micCN, micCP, micNP, AG_ase, BG_ase, BP_ase, CE_ase, P_ase, N_ase, S_ase, XY_ase, LP_ase, PX_ase, CO2, enzCN, enzCP, enzNP# REMOVED soil prop: micN, micNP, enzCN, enzCP, BP_ase, CE_ase, LP_ase, N_ase, P_ase# Remaining soil prop: micC, micP, micCN, micCP, AG_ase, BG_ase, S_ase, XY_ase, PX_ase, CO2, enzNP# 11 total, only works with 13# capscale Removed NONE``````{r}soil_funct_cap <-capscale(tmp_comm ~ micC + micP + micCN + micCP + AG_ase + BG_ase + S_ase + XY_ase + PX_ase + CO2 + enzNP, tmp_md, dist ="bray")``````{r}#| echo: false#| eval: truesoil_funct_cap```Now we can look at the variance against each principal component. ```{r}#| echo: false#| eval: truescreeplot(soil_funct_cap)```And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The `ggplot` function `autoplot` stores these data in a more accessible way than the raw results from `capscale````{r}#| eval: truebase::plot(soil_funct_cap) tmp_auto_plt <-autoplot(soil_funct_cap, arrows =TRUE)tmp_auto_plt``````{r}#| echo: falseanova(soil_funct_cap) # overall test of the significant of the analysisanova(soil_funct_cap, by ="axis", perm.max=500) # test axes for significanceanova(soil_funct_cap, by ="terms", permu=500) # test for sign. environ. variables```Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function `scores` which gets species or site scores from the ordination.```{r}#| code-fold: truetmp_samp_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="sites")tmp_samp_scores[,1] <-NULLtmp_samp_scores <- tmp_samp_scores %>% dplyr::rename(SampleID = Label)tmp_md_sub <- tmp_md[, 1:4]tmp_md_sub <- tmp_md_sub %>% tibble::rownames_to_column("SampleID")soil_funct_plot_data <- dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")```Now we have a new data frame that contains sample details and capscale values. ```{r}#| echo: false#| eval: truesoil_funct_plot_data```We can then do the same with the metadata vectors. Here though we only need the scores and parameter name. ```{r}#| code-fold: truesoil_funct_md_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="biplot")soil_funct_md_scores[,1] <-NULLsoil_funct_md_scores <- soil_funct_md_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")``````{r}#| echo: false#| eval: truesoil_funct_md_scores```Let's run some quick correlations of metadata with ordination axes to see which parameters are significant. For this we use the vegan function `envfit`.```{r}#| code-fold: truetmp_samp_scores_sub <- soil_funct_plot_data[, 6:7]tmp_samp_scores_sub <-as.matrix(tmp_samp_scores_sub)tmp_param_list <- soil_funct_md_scores$parameterstmp_md_sub <-subset(tmp_md, select = tmp_param_list)envfit_soil_funct_md <-envfit(tmp_samp_scores_sub, tmp_md_sub,perm =1000, choices =c(1, 2))``````{r}#| echo: false#| eval: trueenvfit_soil_funct_md``````{r}#| code-fold: truesoil_funct_md_signif_hits <- base::subset(envfit_soil_funct_md$vectors$pvals, c(envfit_soil_funct_md$vectors$pvals <0.05& envfit_soil_funct_md$vectors$r >0.4))soil_funct_md_signif_hits <-data.frame(soil_funct_md_signif_hits)soil_funct_md_signif_hits <-rownames(soil_funct_md_signif_hits)soil_funct_md_signif <- soil_funct_md_scores[soil_funct_md_scores$parameters %in% soil_funct_md_signif_hits,]soil_funct_md_signif$parameters````envfit` found that `r soil_funct_md_signif$parameters` were significantly correlated. Now let's see if the same parameters are significant for the `envfit` and `bioenv` analyses.```{r}#| echo: false#| eval: true#| results: hold#| comment: '' print("Significant parameters from bioenv analysis.")row.names(summary(soil_funct_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")soil_funct_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(soil_funct_bioenv_ind_mantel)), soil_funct_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(soil_funct_md_signif$parameters, row.names(summary(soil_funct_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")soil_funct_sig_diff <- base::union(soil_funct_md_signif$parameters, row.names(summary(soil_funct_bioenv_ind_mantel)))soil_funct_sig_diff``````{r}#| code-fold: true#new_soil_funct_md_signif_hits <- append(soil_funct_md_signif_hits, soil_funct_sig_diff)new_soil_funct_md_signif_hits <- soil_funct_sig_diffsoil_funct_md_signif_all <- soil_funct_md_scores[soil_funct_md_scores$parameters %in% new_soil_funct_md_signif_hits,]```Check. Next, we run `envfit` for the ASVs.```{r}#| code-fold: trueenvfit_soil_funct_asv <-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10], perm =1000, choices =c(1, 2))soil_funct_asv_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="species")soil_funct_asv_scores <- soil_funct_asv_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")soil_funct_asv_scores[,1] <-NULL``````{r}#| echo: false#| eval: trueenvfit_soil_funct_asv``````{r}#| code-fold: truesoil_funct_asv_signif_hits <- base::subset(envfit_soil_funct_asv$vectors$pvals, c(envfit_soil_funct_asv$vectors$pvals <0.05& envfit_soil_funct_asv$vectors$r >0.5))soil_funct_asv_signif_hits <-data.frame(soil_funct_asv_signif_hits)soil_funct_asv_signif_hits <-rownames(soil_funct_asv_signif_hits)soil_funct_asv_signif <- soil_funct_asv_scores[soil_funct_asv_scores$parameters %in% soil_funct_asv_signif_hits,]``````{r}#| echo: false#| eval: truesoil_funct_asv_signif``````{r}#| code-fold: truesoil_funct_md_signif_all$variable_type <-"metadata"soil_funct_asv_signif$variable_type <-"ASV"soil_funct_bioplot_data <-rbind(soil_funct_md_signif_all, soil_funct_asv_signif)```The last thing to do is categorize parameters scores and ASV scores into different variable types for plotting.```{r}#| code-fold: truesoil_funct_bioplot_data_md <-subset(soil_funct_bioplot_data, soil_funct_bioplot_data$variable_type =="metadata")soil_funct_bioplot_data_asv <-subset(soil_funct_bioplot_data, soil_funct_bioplot_data$variable_type =="ASV")```<details><summary>Show the code for the plot</summary>```{r}soil_funct_cap_vals <-data.frame(soil_funct_cap$CCA$eig[1:2])soil_funct_cap1 <-signif((soil_funct_cap_vals[1,] *100), digits=3)soil_funct_cap2 <-signif((soil_funct_cap_vals[2,] *100), digits=3)cpa1_lab <-paste("CAP1", " (", soil_funct_cap1, "%)", sep ="")cpa2_lab <-paste("CAP2", " (", soil_funct_cap2, "%)", sep ="")swel_col <-c("#2271B2", "#71B222", "#B22271")soil_funct_plot <-ggplot(soil_funct_plot_data) +geom_point(mapping =aes(x = CAP1, y = CAP2, shape = TREAT,colour = TREAT_T), size =4) +scale_colour_manual(values = swel_col) +# geom_text(data = soil_funct_plot_data, aes(x = CAP1, y = CAP2, #UNCOMMENT to add sample labels# label = SampleID), size = 3) + geom_segment(aes(x =0, y =0, xend = CAP1, yend = CAP2),data = soil_funct_bioplot_data_md, linetype ="solid",arrow =arrow(length =unit(0.3, "cm")), size =0.4, color ="#191919") +geom_text(data = soil_funct_bioplot_data_md, aes(x = CAP1, y = CAP2, label = parameters), size =3,nudge_x =0.1, nudge_y =0.05) +### This code adds ASV vectors, Uncomment to add#geom_segment(aes(x = 0, y = 0, xend = CAP1, yend = CAP2),# data = soil_funct_bioplot_data_asv, linetype = "solid",# arrow = arrow(length = unit(0.3, "cm")), size = 0.2,# color = "#676767") +#geom_text(data = soil_funct_bioplot_data_asv, # aes(x = CAP1, y = CAP2, label = parameters), size = 2.5,# nudge_x = 0.05, nudge_y = 0.05) +theme_classic(base_size =12) +labs(title ="Capscale Analysis",subtitle ="Soil Functional Response", x = cpa1_lab, y = cpa2_lab)soil_funct_plot <- soil_funct_plot +coord_fixed() +theme(aspect.ratio=1)soil_funct_plotpng("files/metadata/figures/ssu18_soil_funct_capscale.png",height =16, width =20, units ='cm', res =600, bg ="white")soil_funct_plotinvisible(dev.off())pdf("files/metadata/figures/ssu18_soil_funct_capscale.pdf",height =5, width =6)soil_funct_plotdev.off()```</details>```{r}#| echo: falserm(list =ls(pattern ="tmp_"))```### Temperature Adaptation```{r}#| code-fold: truetmp_md <- ssu18_select_mc_norm_split_no_ac$temp_adapttmp_md$TREAT_T <-as.character(tmp_md$TREAT_T)tmp_comm <-data.frame(t(ssu18_select_mc_norm_split_no_ac$data_loaded))temp_adapt_rank <-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow","bra", "kul"), stepacross =FALSE, method ="spearman")``````{r}#| echo: false#| eval: truetemp_adapt_rank```Let's run `capscale` using Bray-Curtis. Note, we have `r ncol(ssu18_select_mc_norm_split_no_ac$temp_adapt) - 8` metadata parameters in this group but, for some reason, `capscale` only works with 13 parameters. This may have to do with degrees of freedom? * Starting properties: AG_Q10, BG_Q10, BP_Q10, CE_Q10, P_Q10, N_Q10, S_Q10, XY_Q10, LP_Q10, PX_Q10, CUEcn, CUEcp, NUE, PUE, Tmin, SI* Autocorrelated removed: NUE, PUE, SI* Remove for capscale: NONE```{r}#| echo: false# All temp_adapt: AG_Q10, BG_Q10, BP_Q10, CE_Q10, P_Q10, N_Q10, S_Q10, XY_Q10, LP_Q10, PX_Q10, CUEcn, CUEcp, NUE, PUE, Tmin, SI# REMOVED temp_adapt: NUE, PUE, SI# Remaining temp_adapt: AG_Q10, BG_Q10, BP_Q10, CE_Q10, P_Q10, N_Q10, S_Q10, XY_Q10, LP_Q10, PX_Q10, CUEcn, CUEcp, Tmin# 14 total, only works with 13# Removed: NONE``````{r}temp_adapt_cap <-capscale(tmp_comm ~ AG_Q10 + BG_Q10 + BP_Q10 + CE_Q10 + P_Q10 + N_Q10 + S_Q10 + XY_Q10 + LP_Q10 + PX_Q10 + CUEcn + CUEcp + Tmin, tmp_md, dist ="bray")``````{r}#| echo: false#| eval: truetemp_adapt_cap```Now we can look at the variance against each principal component. ```{r}#| echo: false#| eval: truescreeplot(temp_adapt_cap)```And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The `ggplot` function `autoplot` stores these data in a more accessible way than the raw results from `capscale````{r}#| eval: truebase::plot(temp_adapt_cap) tmp_auto_plt <-autoplot(temp_adapt_cap, arrows =TRUE)tmp_auto_plt``````{r}#| echo: falseanova(temp_adapt_cap) # overall test of the significant of the analysisanova(temp_adapt_cap, by ="axis", perm.max =500) # test axes for significanceanova(temp_adapt_cap, by ="terms", permu =500) # test for sign. environ. variables```Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function `scores` which gets species or site scores from the ordination.```{r}#| code-fold: truetmp_samp_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="sites")tmp_samp_scores[,1] <-NULLtmp_samp_scores <- tmp_samp_scores %>% dplyr::rename(SampleID = Label)tmp_md_sub <- tmp_md[, 1:4]tmp_md_sub <- tmp_md_sub %>% tibble::rownames_to_column("SampleID")temp_adapt_plot_data <- dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")```Now we have a new data frame that contains sample details and capscale values. ```{r}#| echo: false#| eval: truetemp_adapt_plot_data```We can then do the same with the metadata vectors. Here though we only need the scores and parameter name. ```{r}#| code-fold: truetemp_adapt_md_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="biplot")temp_adapt_md_scores[,1] <-NULLtemp_adapt_md_scores <- temp_adapt_md_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")``````{r}#| echo: false#| eval: truetemp_adapt_md_scores```Let's run some quick correlations of metadata with ordination axes to see which parameters are significant. For this we use the vegan function `envfit`.```{r}#| code-fold: truetmp_samp_scores_sub <- temp_adapt_plot_data[, 6:7]tmp_samp_scores_sub <-as.matrix(tmp_samp_scores_sub)tmp_param_list <- temp_adapt_md_scores$parameterstmp_md_sub <-subset(tmp_md, select = tmp_param_list)envfit_temp_adapt_md <-envfit(tmp_samp_scores_sub, tmp_md_sub,perm =1000, choices =c(1, 2))``````{r}#| echo: false#| eval: trueenvfit_temp_adapt_md``````{r}#| code-fold: truetemp_adapt_md_signif_hits <- base::subset(envfit_temp_adapt_md$vectors$pvals, c(envfit_temp_adapt_md$vectors$pvals <0.05& envfit_temp_adapt_md$vectors$r >0.4))temp_adapt_md_signif_hits <-data.frame(temp_adapt_md_signif_hits)temp_adapt_md_signif_hits <-rownames(temp_adapt_md_signif_hits)temp_adapt_md_signif <- temp_adapt_md_scores[temp_adapt_md_scores$parameters %in% temp_adapt_md_signif_hits,]````envfit` found that `r temp_adapt_md_signif$parameters` were significantly correlated. Now let's see if the same parameters are significant for the `envfit` and `bioenv` analyses.```{r}#| echo: false#| eval: true#| results: hold#| comment: '' print("Significant parameters from bioenv analysis.")row.names(summary(temp_adapt_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")temp_adapt_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(temp_adapt_bioenv_ind_mantel)), temp_adapt_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(temp_adapt_md_signif$parameters, row.names(summary(temp_adapt_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")temp_adapt_sig_diff <- base::union(temp_adapt_md_signif$parameters, row.names(summary(temp_adapt_bioenv_ind_mantel)))temp_adapt_sig_diff``````{r}#| code-fold: true#new_temp_adapt_md_signif_hits <- base::append(temp_adapt_md_signif_hits, temp_adapt_sig_diff)new_temp_adapt_md_signif_hits <- temp_adapt_sig_difftemp_adapt_md_signif_all <- temp_adapt_md_scores[temp_adapt_md_scores$parameters %in% new_temp_adapt_md_signif_hits,]```Check. Next, we run `envfit` for the ASVs.```{r}#| code-fold: trueenvfit_temp_adapt_asv <-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10],perm =1000, choices =c(1, 2))temp_adapt_asv_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="species")temp_adapt_asv_scores <- temp_adapt_asv_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")temp_adapt_asv_scores[,1] <-NULL``````{r}#| echo: false#| eval: trueenvfit_temp_adapt_asv``````{r}#| code-fold: truetemp_adapt_asv_signif_hits <- base::subset(envfit_temp_adapt_asv$vectors$pvals, c(envfit_temp_adapt_asv$vectors$pvals <0.05& envfit_temp_adapt_asv$vectors$r >0.5))temp_adapt_asv_signif_hits <-data.frame(temp_adapt_asv_signif_hits)temp_adapt_asv_signif_hits <-rownames(temp_adapt_asv_signif_hits)temp_adapt_asv_signif <- temp_adapt_asv_scores[temp_adapt_asv_scores$parameters %in% temp_adapt_asv_signif_hits,]``````{r}#| echo: false#| eval: truetemp_adapt_asv_signif``````{r}#| code-fold: truetemp_adapt_md_signif_all$variable_type <-"metadata"temp_adapt_asv_signif$variable_type <-"ASV"temp_adapt_bioplot_data <-rbind(temp_adapt_md_signif_all, temp_adapt_asv_signif)```The last thing to do is categorize parameters scores and ASV scores into different variable types for plotting.```{r}#| code-fold: truetemp_adapt_bioplot_data_md <-subset(temp_adapt_bioplot_data, temp_adapt_bioplot_data$variable_type =="metadata")temp_adapt_bioplot_data_asv <-subset(temp_adapt_bioplot_data, temp_adapt_bioplot_data$variable_type =="ASV")```<details><summary>Show the code for the plot</summary>```{r}temp_adapt_cap_vals <-data.frame(temp_adapt_cap$CCA$eig[1:2])temp_adapt_cap1 <-signif((temp_adapt_cap_vals[1,] *100), digits=3)temp_adapt_cap2 <-signif((temp_adapt_cap_vals[2,] *100), digits=3)cpa1_lab <-paste("CAP1", " (", temp_adapt_cap1, "%)", sep ="")cpa2_lab <-paste("CAP2", " (", temp_adapt_cap2, "%)", sep ="")swel_col <-c("#2271B2", "#71B222", "#B22271")temp_adapt_plot <-ggplot(temp_adapt_plot_data) +geom_point(mapping =aes(x = CAP1, y = CAP2, shape = TREAT,colour = TREAT_T), size =4) +scale_colour_manual(values = swel_col) +# geom_text(data = temp_adapt_plot_data, aes(x = CAP1, y = CAP2, #UNCOMMENT to add sample labels# label = SampleID), size = 3) + geom_segment(aes(x =0, y =0, xend = CAP1, yend = CAP2),data = temp_adapt_bioplot_data_md, linetype ="solid",arrow =arrow(length =unit(0.3, "cm")), size =0.4,color ="#191919", inherit.aes =FALSE) +geom_text(data = temp_adapt_bioplot_data_md, aes(x = CAP1, y = CAP2, label = parameters), size =3,nudge_x =0.1, nudge_y =0.05) +### This code adds ASV vectors, Uncomment to add# geom_segment(aes(x = 0, y = 0, xend = CAP1, yend = CAP2),# data = temp_adapt_bioplot_data_asv, linetype = "solid",# arrow = arrow(length = unit(0.3, "cm")), size = 0.2,# color = "#676767") +# geom_text(data = temp_adapt_bioplot_data_asv, # aes(x = CAP1, y = CAP2, label = parameters), size = 2.5,# nudge_x = 0.05, nudge_y = 0.05) +theme_classic(base_size =12) +labs(title ="Capscale Analysis",subtitle ="Temperature Adaptation",x = cpa1_lab, y = cpa2_lab)temp_adapt_plot <- temp_adapt_plot +coord_fixed() +theme(aspect.ratio=1)temp_adapt_plotpng("files/metadata/figures/ssu18_temp_adapt_capscale.png",height =16, width =20, units ='cm', res =600, bg ="white")temp_adapt_plotinvisible(dev.off())pdf("files/metadata/figures/ssu18_temp_adapt_capscale.pdf",height =5, width =6)temp_adapt_plotdev.off()```</details>```{r}#| echo: falserm(list =ls(pattern ="tmp_"))```## Capscale Plots::: {.column-body}::: {.panel-tabset}#### Edaphic properties```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5edaphic_plot```<small>`r caption_fig_ssu("ssu_cap_edaph_plots")`</small>#### Soil Functional Response```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5soil_funct_plot```<small>`r caption_fig_ssu("ssu_cap_funct_plots")`</small>#### Temperature Adaptation```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5temp_adapt_plot```<small>`r caption_fig_ssu("ssu_cap_temp_plots")`</small>::::::```{r}#| echo: falsessu18_capscale_plots <-ggarrange( edaphic_plot, soil_funct_plot, temp_adapt_plot,ncol =3, nrow =1, common.legend =TRUE, legend ="bottom")ssu18_capscale_plotsdev.off()png("files/metadata/figures/ssu18_capscale_plots.png", height =12, width =36,units ='cm', res =600, bg ="white")ssu18_capscale_plotsdev.off()pdf("files/metadata/figures/ssu18_capscale_plots.pdf", height =6, width =18)ssu18_capscale_plotsdev.off()``````{r}#| echo: false################################################### CODE FOR SUMMARY DATA ######################################################### Edaphicbioenv_set <- edaphic_bioenv_ind_mantelenvfit_set <- envfit_edaphic_mdres_name <-"ssu18_edaphic_sig_summary"metadata_set <-"edaphic"## BIOENVbioenv_tab <-NULLfor (i innames(bioenv_set)) { tmp_r <-data.frame(bioenv_set[[i]]$statistic)row.names(tmp_r) <- i tmp_p <-data.frame(bioenv_set[[i]]$signif)row.names(tmp_p) <- i bioenv_tab <-rbind(bioenv_tab, data.frame(tmp_r, tmp_p))rm(list =ls(pattern ="tmp_"))}bioenv_tab <- bioenv_tab %>% dplyr::rename("r (bioenv)"=1, "pval (bioenv)"=2) %>% tibble::rownames_to_column("parameter")## ENVFITtmp_r <-data.frame(envfit_set$vectors$r)tmp_r <- tmp_r %>% tibble::rownames_to_column("parameter")tmp_p <-data.frame(envfit_set$vectors$pvals)tmp_p <- tmp_p %>% tibble::rownames_to_column("parameter")tmp_rp <- dplyr::full_join(tmp_r, tmp_p, by ="parameter")tmp_rp <- tmp_rp %>% dplyr::rename("r2 (envfit)"=2, "Pr(>r) (envfit)"=3)tmp_rp <- base::subset(tmp_rp, c(tmp_rp$`Pr(>r)`<0.05& tmp_rp$r2 >0.4))tmp_summary <- dplyr::full_join(tmp_rp, bioenv_tab, by ="parameter") tmp_summary$metadata_set <- metadata_settmp_summary <- tmp_summary[, c(6,1:5)]assign(res_name, tmp_summary)rm(list =ls(pattern ="tmp_"))``````{r}#| echo: false## SOIL_FUNCTbioenv_set <- soil_funct_bioenv_ind_mantelenvfit_set <- envfit_soil_funct_mdres_name <-"ssu18_soil_funct_sig_summary"metadata_set <-"functional_response"## BIOENVbioenv_tab <-NULLfor (i innames(bioenv_set)) { tmp_r <-data.frame(bioenv_set[[i]]$statistic)row.names(tmp_r) <- i tmp_p <-data.frame(bioenv_set[[i]]$signif)row.names(tmp_p) <- i bioenv_tab <-rbind(bioenv_tab, data.frame(tmp_r, tmp_p))rm(list =ls(pattern ="tmp_"))}bioenv_tab <- bioenv_tab %>% dplyr::rename("r (bioenv)"=1, "pval (bioenv)"=2) %>% tibble::rownames_to_column("parameter")## ENVFITtmp_r <-data.frame(envfit_set$vectors$r)tmp_r <- tmp_r %>% tibble::rownames_to_column("parameter")tmp_p <-data.frame(envfit_set$vectors$pvals)tmp_p <- tmp_p %>% tibble::rownames_to_column("parameter")tmp_rp <- dplyr::full_join(tmp_r, tmp_p, by ="parameter")tmp_rp <- tmp_rp %>% dplyr::rename("r2 (envfit)"=2, "Pr(>r) (envfit)"=3)tmp_rp <- base::subset(tmp_rp, c(tmp_rp$`Pr(>r)`<0.05& tmp_rp$r2 >0.4))tmp_summary <- dplyr::full_join(tmp_rp, bioenv_tab, by ="parameter") tmp_summary$metadata_set <- metadata_settmp_summary <- tmp_summary[, c(6,1:5)]assign(res_name, tmp_summary)rm(list =ls(pattern ="tmp_"))``````{r}#| echo: false## TEMPbioenv_set <- temp_adapt_bioenv_ind_mantelenvfit_set <- envfit_temp_adapt_mdres_name <-"ssu18_temp_adapt_sig_summary"metadata_set <-"temperature adaptation"## BIOENVbioenv_tab <-NULLfor (i innames(bioenv_set)) { tmp_r <-data.frame(bioenv_set[[i]]$statistic)row.names(tmp_r) <- i tmp_p <-data.frame(bioenv_set[[i]]$signif)row.names(tmp_p) <- i bioenv_tab <-rbind(bioenv_tab, data.frame(tmp_r, tmp_p))rm(list =ls(pattern ="tmp_"))}bioenv_tab <- bioenv_tab %>% dplyr::rename("r (bioenv)"=1, "pval (bioenv)"=2) %>% tibble::rownames_to_column("parameter")## ENVFITtmp_r <-data.frame(envfit_set$vectors$r)tmp_r <- tmp_r %>% tibble::rownames_to_column("parameter")tmp_p <-data.frame(envfit_set$vectors$pvals)tmp_p <- tmp_p %>% tibble::rownames_to_column("parameter")tmp_rp <- dplyr::full_join(tmp_r, tmp_p, by ="parameter")tmp_rp <- tmp_rp %>% dplyr::rename("r2 (envfit)"=2, "Pr(>r) (envfit)"=3)tmp_rp <- base::subset(tmp_rp, c(tmp_rp$`Pr(>r)`<0.05& tmp_rp$r2 >0.4))tmp_summary <- dplyr::full_join(tmp_rp, bioenv_tab, by ="parameter") tmp_summary$metadata_set <- metadata_settmp_summary <- tmp_summary[, c(6,1:5)]assign(res_name, tmp_summary)rm(list =ls(pattern ="tmp_"))rm(bioenv_set, envfit_set, res_name, metadata_set, bioenv_tab)ssu18_sig_summary <-rbind(ssu18_edaphic_sig_summary, ssu18_soil_funct_sig_summary, ssu18_temp_adapt_sig_summary)rm(ssu18_edaphic_sig_summary, ssu18_soil_funct_sig_summary, ssu18_temp_adapt_sig_summary)``````{r}#| echo: falsesave.image("page_build/metadata_ssu18_wf.rdata")gdata::keep(metad, ssu18_sig_summary, sure =TRUE)objects()```# ITS```{r}#| include: false#| eval: true## Load to build page only #2#remove(list = ls())load("page_build/metadata_its18_wf.rdata")```## Data Formating ```{r}#| include: false## Initial Load for ANALYSIS #1#remove(list = ls())set.seed(119)its18_ps_work <-readRDS("files/alpha/rdata/its18_ps_work.rds")its18_ps_filt <-readRDS("files/alpha/rdata/its18_ps_filt.rds")its18_ps_perfect <-readRDS("files/alpha/rdata/its18_ps_perfect.rds")its18_ps_pime <-readRDS("files/alpha/rdata/its18_ps_pime.rds")```1. Transform `ps` ObjectsThe first step is to transform the phyloseq objects. ```{r}#| code-fold: truesamp_ps <-c("its18_ps_work", "its18_ps_pime", "its18_ps_perfect", "its18_ps_work_otu", "its18_ps_pime_otu", "its18_ps_perfect_otu")for (i in samp_ps) { tmp_get <-get(i) tmp_ps <-transform_sample_counts(tmp_get, function(otu) 1e5* otu/sum(otu)) tmp_ps@phy_tree <-NULL tmp_ps <-prune_samples(sample_sums(tmp_ps) >0, tmp_ps) tmp_samp <-data.frame(sample_data(tmp_get)) tmp_samp <- tmp_samp %>% dplyr::rename("TREAT_T"="TEMP") tmp_ps <-merge_phyloseq(tmp_ps, sample_data)sample_data(tmp_ps) <- tmp_samp tmp_name <- purrr::map_chr(i, ~paste0(., "_prop"))print(tmp_name)assign(tmp_name, tmp_ps)rm(list =ls(pattern ="tmp_"))}```2. Format for `mctoolsr`Then a bit of formatting to make the data compatible with `mctoolsr`. ```{r}#| code-fold: truefor (i in samp_ps) { tmp_get <-get(purrr::map_chr(i, ~paste0(., "_prop"))) tmp_tax <-data.frame(tax_table(tmp_get)) tmp_tax$ASV_SEQ <-NULL tmp_col_names <-colnames(tmp_tax) tmp_tax_merge <- tmp_tax %>% tidyr::unite(taxonomy, all_of(tmp_col_names), sep =";") tmp_tax_merge <- tmp_tax_merge %>% tibble::rownames_to_column("#OTU_ID") tmp_otu <-data.frame(t(otu_table(tmp_get))) tmp_otu <- tmp_otu %>% tibble::rownames_to_column("#OTU_ID") tmp_otu_tax <- dplyr::left_join(tmp_otu, tmp_tax_merge, by ="#OTU_ID") tmp_samp <-data.frame(sample_data(tmp_get)) tmp_samp[,c(1,3)] <-NULL tmp_samp <- tmp_samp %>% tibble::rownames_to_column("#SampleID") tmp_metad <- metad %>% dplyr::rename("#SampleID"="id") tmp_metad[,2:5] <-NULL tmp_md <- dplyr::left_join(tmp_samp, tmp_metad, by ="#SampleID") tmp_otu_name <- purrr::map_chr(i, ~paste0(., "_otu_tax"))print(tmp_otu_name)assign(tmp_otu_name, tmp_otu_tax) tmp_md_name <- purrr::map_chr(i, ~paste0(., "_md"))print(tmp_md_name)assign(tmp_md_name, tmp_md) tmp_path <-file.path("files/metadata/tables/")write_delim(tmp_otu_tax, paste(tmp_path, tmp_otu_name, ".txt", sep =""), delim ="\t")write_delim(tmp_md, paste(tmp_path, tmp_md_name, ".txt", sep =""), delim ="\t")rm(list =ls(pattern ="tmp_"))}``````{r}#| code-fold: truefor (i in samp_ps) { tmp_path <-file.path("files/metadata/tables/") tmp_otu_name <- purrr::map_chr(i, ~paste0(., "_otu_tax")) tmp_md_name <- purrr::map_chr(i, ~paste0(., "_md")) tmp_tax_table_fp <-paste(tmp_path, tmp_otu_name, ".txt", sep ="") tmp_map_fp <-paste(tmp_path, tmp_md_name, ".txt", sep ="") tmp_input <-load_taxa_table(tmp_tax_table_fp, tmp_map_fp) tmp_input_name <- purrr::map_chr(i, ~paste0(., "_mc"))print(tmp_input_name)assign(tmp_input_name, tmp_input)rm(list =ls(pattern ="tmp_"))}rm(list =ls(pattern ="_md"))rm(list =ls(pattern ="_otu_tax"))```## Choose Data SetAt this point in the code we need to choose a data set to use, formatted with `mctoolsr`. Remember, there are four choices:1) Complete ASV data set.2) Arbitrary filtered ASV data set.3) PERfect filtered ASV data set.4) PIME filtered ASV data set.This way, if we want to test other data sets we only need to change the name here.```{r}objects(pattern ="_mc")its18_select_mc <- its18_ps_pime_mc```## Normality TestsBefore proceeding, we need to test each parameter in the metadata to see which ones are and are not normally distributed. For that, we use the Shapiro-Wilk Normality Test. Here we only need one of the metadata files.```{r}#| code-fold: truetemp_md <- its18_select_mc$map_loadedtemp_md[,1:9] <-NULLshap_results <-NULLfor (i incolnames(temp_md)) { tmp_shap <-shapiro.test(temp_md[[i]]) tmp_p <-round(tmp_shap$p.value, digits =5) tmp_res <-eval(isTRUE(tmp_shap$p.value <0.05)) shap_results <-rbind(shap_results, data.frame(i, tmp_p, tmp_res))rm(list =ls(pattern ="tmp_"))}colnames(shap_results) <-c("parameter", "p-value", "tranform")shap_resultsdplyr::filter(shap_results, tranform =="TRUE")md_to_tranform <- shap_results$parameter[shap_results$tranform ==TRUE]rm(list =ls(pattern ="temp_md"))```<details><summary>**Show the results** of each normality test for metadata parameters</summary></br><small>`r caption_tab_its("its_normality_tests")`</small>```{r}#| echo: false#| eval: trueseq_table <- shap_resultsreactable(seq_table,defaultColDef =colDef(headerStyle =list(fontSize ="0.8em"),header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =1),align ="center", filterable =TRUE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold") ), columns =list(parameter =colDef(name ="Parameter", sticky ="left", style =list(borderRight ="1px solid #eee"),headerStyle =list(borderRight ="1px solid #eee", fontSize ="0.9em"), align ="left",minWidth =150), `p-value`=colDef(name ="p-value", style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color, fontWeight ="bold")}) ), searchable =TRUE, defaultPageSize =60, pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =FALSE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =FALSE, wrap =FALSE, showSortable =TRUE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```</details>Looks like we need to transform `r length(md_to_tranform)` metadata parameters.## Normalize ParametersHere we use the R package [`bestNormalize`](https://cran.r-project.org/web/packages/bestNormalize/vignettes/bestNormalize.html) to find and execute the best normalizing transformation. The function will test the following normalizing transformations:- `arcsinh_x` performs an arcsinh transformation.- `boxcox` Perform a Box-Cox transformation and center/scale a vector to attempt normalization. `boxcox` estimates the optimal value of lambda for the Box-Cox transformation. The function will return an error if a user attempt to transform nonpositive data.- `yeojohnson` Perform a Yeo-Johnson Transformation and center/scale a vector to attempt normalization. `yeojohnson` estimates the optimal value of lambda for the Yeo-Johnson transformation. The Yeo-Johnson is similar to the Box-Cox method, however it allows for the transformation of nonpositive data as well. - `orderNorm` The Ordered Quantile (ORQ) normalization transformation, `orderNorm()`, is a rank-based procedure by which the values of a vector are mapped to their percentile, which is then mapped to the same percentile of the normal distribution. Without the presence of ties, this essentially guarantees that the transformation leads to a uniform distribution.- `log_x` performs a simple log transformation. The parameter a is essentially estimated by the training set by default (estimated as the minimum possible to some extent epsilon), while the base must be specified beforehand. The default base of the log is 10.- `sqrt_x` performs a simple square-root transformation. The parameter a is essentially estimated by the training set by default (estimated as the minimum possible), while the base must be specified beforehand.- `exp_x` performs a simple exponential transformation.See this GitHub issue ([#5](https://github.com/petersonR/bestNormalize/issues/5)) for a description on getting reproducible results. Apparently, you can get different results because the `bestNormalize()` function uses repeated cross-validation (and doesn't automatically set the seed), so the results will be slightly different each time the function is executed.```{r}#| code-fold: trueset.seed(119)for (i in md_to_tranform) { tmp_md <- its18_select_mc$map_loaded tmp_best_norm <-bestNormalize(tmp_md[[i]], r =1, k =5, loo =TRUE) tmp_name <- purrr::map_chr(i, ~paste0(., "_best_norm_test"))assign(tmp_name, tmp_best_norm)print(tmp_name)rm(list =ls(pattern ="tmp_"))}```<details><summary>Show the chosen transformations</summary>```{r}#| echo: false#| eval: truefor (i in md_to_tranform) { tmp_get <-get(purrr::map_chr(i, ~paste0(., "_best_norm_test"))) tmp_print <-c("bestNormalize Chosen transformation of", i)cat("\n")cat("##", tmp_print, "##", "\n")print(tmp_get$chosen_transform)cat("_____________________________________")cat("\n")rm(list =ls(pattern ="tmp_"))}```</details><details><summary>Show the complete `bestNormalize` results</summary>```{r}#| echo: false#| eval: truefor (i in md_to_tranform) { tmp_get <-get(purrr::map_chr(i, ~paste0(., "_best_norm_test"))) tmp_print <-c("Results of bestNormalize for", i)cat("\n")cat("##", tmp_print, "##", "\n")print(tmp_get)cat("_____________________________________")cat("\n")rm(list =ls(pattern ="tmp_"))}```</details>Great, now we can add the normalized transformed data back to our `mctoolsr` metadata file. ```{r}#| code-fold: trueits18_select_mc_norm <- its18_select_mcfor (i in md_to_tranform) { tmp_get <-get(purrr::map_chr(i, ~paste0(i, "_best_norm_test"))) tmp_new_data <- tmp_get$x.t its18_select_mc_norm$map_loaded[[i]] <- tmp_new_datarm(list =ls(pattern ="tmp_"))} ```And rerun the Shapiro Tests. ```{r}#| code-fold: truetemp_md_norm <- its18_select_mc_norm$map_loadedtemp_md_norm[,1:9] <-NULLshap_results_norm <-NULLfor (i incolnames(temp_md_norm)) { tmp_shap <-shapiro.test(temp_md_norm[[i]]) tmp_p <-round(tmp_shap$p.value, digits =5) tmp_res <-eval(isTRUE(tmp_shap$p.value <0.05)) shap_results_norm <-rbind(shap_results_norm, data.frame(i, tmp_p, tmp_res))rm(list =ls(pattern ="tmp_"))}colnames(shap_results_norm) <-c("parameter", "p-value", "tranform")shap_results_normrm(list =ls(pattern ="temp_md_norm"))```And check if there are any parameters that are still significant for the normality test.```{r}#| eval: trueshap_results$parameter[shap_results_norm$tranform ==TRUE]```Ok. Looks like `bestNormalize` was unable to find a suitable transformation for `Al` and `Fe`. This is likely because there is very little variation in these metadata and/or there are too few significant digits.## Normalized MetadataFinally, here is a new summary table that includes all of the normalized data.</br>```{r}#| echo: false#| eval: truemetad_norm <- its18_select_mc_norm$map_loadedmetad_norm <- metad_norm %>% tibble::rownames_to_column("Sample_ID")metad_norm <- metad_norm[order(match(metad_norm$Sample_ID, metad$Sample_ID)), ]metad_norm[,2:9] <-NULLseq_table <- metad_normwrite.table(metad_norm, "files/metadata/tables/its18_normalized_metadata.txt", row.names =FALSE, sep ="\t", quote =FALSE)```<small>`r caption_tab_its("its_normalized_metadata")`</small>```{r}#| echo: false#| eval: trueseq_table <- seq_table %>% dplyr::mutate(across(.cols =c(3:5, 7:9, 14:15,19:20, 24,26,30,34:38,40, 42:43, 49, 52:53,55,57,60:62), round, digits =3)) reactable(seq_table,defaultColDef =colDef(headerStyle =list(fontSize ="0.8em"),header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =1),align ="center", filterable =TRUE, sortable =TRUE, resizable =TRUE,footerStyle =list(fontWeight ="bold") ), columns =list(Sample_ID =colDef(name ="Sample ID", sticky ="left", style =list(borderRight ="1px solid #eee"),headerStyle =list(borderRight ="1px solid #eee", fontSize ="0.9em"), align ="left",minWidth =150), raw_reads =colDef(footer =function(values) sprintf("%.0f", sum(values))) ), searchable =TRUE, defaultPageSize =5, pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =FALSE, wrap =FALSE, showSortable =TRUE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```{{< downloadthis files/metadata/tables/its18_normalized_metadata.txt dname="its18_normalized_metadata" label="Download table as text file" icon="table" type="primary" >}}```{r}#| echo: false# Code to run individual transformations for each metadata and generate histograms##for (i in md_to_tranform) {## tmp_data <- its18_select_mc$map_loaded## tmp_no_transform_obj <- no_transform(tmp_data[[i]])## tmp_arcsinh_obj <- arcsinh_x(tmp_data[[i]])## #tmp_boxcox_obj <- boxcox(tmp_data[[i]])## tmp_yeojohnson_obj <- yeojohnson(tmp_data[[i]])## tmp_orderNorm_obj <- orderNorm(tmp_data[[i]])## tmp_log_obj <- log_x(tmp_data[[i]])## tmp_sqrt_x_obj <- sqrt_x(tmp_data[[i]])## tmp_exp_x_obj <- exp_x(tmp_data[[i]])## ## tmp_plot_no_transform <- hist(tmp_no_transform_obj$x.t, main = i, breaks = 6, xlab = "no_transform")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_no_transform"))## assign(tmp_plot_name, tmp_plot_no_transform)## print(tmp_plot_name)## ## tmp_plot_arcsinh <- hist(tmp_arcsinh_obj$x.t, main = i, breaks = 6, xlab = "arcsinh")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_arcsinh"))## assign(tmp_plot_name, tmp_plot_arcsinh)## print(tmp_plot_name)## # cannot run because of Al and Fe## #tmp_plot_boxcox <- hist(tmp_boxcox_obj$x.t, main = i, breaks = 6, xlab = "boxcox")## #tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_boxcox"))## #assign(tmp_plot_name, tmp_plot_boxcox)## #print(tmp_plot_name)## ## tmp_plot_yeojohnson <- hist(tmp_yeojohnson_obj$x.t, main = i, breaks = 6, xlab = "yeojohnson")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_yeojohnson"))## assign(tmp_plot_name, tmp_plot_yeojohnson)## print(tmp_plot_name)## ## tmp_plot_orderNorm <- hist(tmp_orderNorm_obj$x.t, main = i, breaks = 6, xlab = "orderNorm")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_orderNorm"))## assign(tmp_plot_name, tmp_plot_orderNorm)## print(tmp_plot_name)## ## tmp_plot_log <- hist(tmp_log_obj$x.t, main = i, breaks = 6, xlab = "log")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_log"))## assign(tmp_plot_name, tmp_plot_log)## print(tmp_plot_name)## ## tmp_plot_sqrt <- hist(tmp_sqrt_x_obj$x.t, main = i, breaks = 6, xlab = "sqrt")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_sqrt"))## assign(tmp_plot_name, tmp_plot_sqrt)## print(tmp_plot_name)## ## tmp_plot_exp <- hist(tmp_exp_x_obj$x.t, main = i, breaks = 6, xlab = "exp")## tmp_plot_name <- purrr::map_chr(i, ~ paste0(., "_hist_plot_exp"))## assign(tmp_plot_name, tmp_plot_exp)## print(tmp_plot_name)#### rm(list = ls(pattern = "tmp_"))##}```## Autocorrelation TestsNext, we test the metadata for autocorrelations. Do we do this on the original data or the transformed data? No idea, so let's do both.### Split MetadataWe need to split the data into different groups.A) Environmental and edaphic propertiesB) Microbial functional responsesC) Temperature adaptation propertiesWe first create lists of metadata parameters. ```{r}#| code-fold: truediv <-c("PLOT", "TREAT", "TREAT_T", "PAIR", "Observed", "Shannon_exp", "InvSimpson", "ATAP")edaphic <-c("AST", "H2O", "N", "P", "Al", "Ca", "Fe", "K", "Mg", "Mn", "Na", "TEB", "ECEC", "pH", "NH4", "NO3", "PO4", "DOC", "DON", "DOCN")soil_funct <-c("micC", "micN", "micP", "micCN", "micCP", "micNP", "AG_ase", "BG_ase", "BP_ase", "CE_ase", "P_ase", "N_ase", "S_ase", "XY_ase", "LP_ase", "PX_ase", "CO2", "enzCN", "enzCP", "enzNP")temp_adapt <-c("AG_Q10", "BG_Q10", "BP_Q10", "CE_Q10", "P_Q10", "N_Q10", "S_Q10", "XY_Q10", "LP_Q10", "PX_Q10", "CUEcn", "CUEcp", "NUE", "PUE", "Tmin", "SI")md_groups <-c("edaphic", "soil_funct", "temp_adapt")# NoT uSed: minPO4, minNH4, minNO3, minTIN```And then use the lists to split the data sets by metadata group. Here, we do this for the original metadata and the metadata after normalization.```{r}#| code-fold: trueselect_md <-c("its18_select_mc", "its18_select_mc_norm")for (i in select_md) {#tmp_get <- get(purrr::map_chr(i, ~ paste0(i, "_mc"))) tmp_get <-get(i) tmp_md_all <- tmp_get$map_loaded tmp_div <- tmp_md_all %>% dplyr::select(all_of(div)) tmp_div <- tmp_div %>% tibble::rownames_to_column("SampleID")## edaphic tmp_sub_edaphic <- tmp_md_all %>% dplyr::select(all_of(edaphic)) tmp_sub_edaphic <- tmp_sub_edaphic %>% tibble::rownames_to_column("SampleID") tmp_edaphic <- dplyr::left_join(tmp_div, tmp_sub_edaphic, by ="SampleID") tmp_edaphic <- tmp_edaphic %>% tibble::column_to_rownames("SampleID")## soil_funct tmp_sub_soil_funct <- tmp_md_all %>% dplyr::select(all_of(soil_funct)) tmp_sub_soil_funct <- tmp_sub_soil_funct %>% tibble::rownames_to_column("SampleID") tmp_soil_funct <- dplyr::left_join(tmp_div, tmp_sub_soil_funct, by ="SampleID") tmp_soil_funct <- tmp_soil_funct %>% tibble::column_to_rownames("SampleID") ## temp_adapt tmp_sub_temp_adapt <- tmp_md_all %>% dplyr::select(all_of(temp_adapt)) tmp_sub_temp_adapt <- tmp_sub_temp_adapt %>% tibble::rownames_to_column("SampleID") tmp_temp_adapt <- dplyr::left_join(tmp_div, tmp_sub_temp_adapt, by ="SampleID") tmp_temp_adapt <- tmp_temp_adapt %>% tibble::column_to_rownames("SampleID") ## combine tmp_list <-list(data_loaded = its18_select_mc$data_loaded, map_loaded = its18_select_mc$map_loaded, taxonomy_loaded = its18_select_mc$taxonomy_loaded,edaphic = tmp_edaphic, soil_funct = tmp_soil_funct, temp_adapt = tmp_temp_adapt) tmp_name <- purrr::map_chr(i, ~paste0(., "_split"))print(tmp_name)assign(tmp_name, tmp_list)rm(list =ls(pattern ="tmp_"))}```### Generate Autocorrelation PlotsA little housekeeping to get rid of parameters we don't need (e.g., plot number, pair, etc.).```{r}#| code-fold: trueedaphic_cor <- its18_select_mc_split$edaphicedaphic_cor[,1:8] <-NULLedaphic_norm_cor <- its18_select_mc_norm_split$edaphicedaphic_norm_cor[,1:8] <-NULLsoil_funct_cor <- its18_select_mc_split$soil_functsoil_funct_cor[,1:8] <-NULLsoil_funct_norm_cor <- its18_select_mc_norm_split$soil_functsoil_funct_norm_cor[,1:8] <-NULLtemp_adapt_cor <- its18_select_mc_split$temp_adapttemp_adapt_cor[,1:8] <-NULLtemp_adapt_norm_cor <- its18_select_mc_norm_split$temp_adapttemp_adapt_norm_cor[,1:8] <-NULL```And finally the code to create the plots. ```{r}#| code-fold: truefor (i inobjects(pattern ="_cor$")) { tmp_get <-get(i) tmp_cormat <-round(cor(tmp_get), 2) tmp_melted_cormat <- reshape2::melt(tmp_cormat) tmp_get_lower_tri <-function(tmp_cormat){ tmp_cormat[upper.tri(tmp_cormat)] <-NAreturn(tmp_cormat) }# Get upper triangle of the correlation matrix tmp_get_upper_tri <-function(tmp_cormat){ tmp_cormat[lower.tri(tmp_cormat)] <-NAreturn(tmp_cormat) } tmp_upper_tri <-tmp_get_upper_tri(tmp_cormat) tmp_melted_cormat <- reshape2::melt(tmp_upper_tri, na.rm =TRUE)ggplot(data = tmp_melted_cormat, aes(x = Var1, y = Var2, fill = value)) +geom_tile() tmp_ggheatmap <-ggplot(data = tmp_melted_cormat, aes(Var2, Var1, fill = value)) +geom_tile(color ="white") +scale_fill_gradient2(low ="blue", high ="red", mid ="white", midpoint =0, limit =c(-1,1), space ="Lab", name="Pearson\nCorrelation") +theme_minimal() +theme(axis.text.x =element_text(angle =45, vjust =1, size =7, hjust =1),axis.text.y =element_text(vjust =1, size =7, hjust =1)) +coord_fixed() +geom_text(aes(Var2, Var1, label = value), color ="black", size =1.75) +theme(axis.title.x =element_blank(),axis.title.y =element_blank(),panel.grid.major =element_blank(),panel.border =element_blank(),panel.background =element_blank(),axis.ticks =element_blank(),legend.justification =c(1, 0),legend.position =c(0.6, 0.7),legend.direction ="horizontal") +guides(fill =guide_colorbar(barwidth =7, barheight =1,title.position ="top", title.hjust =0.5)) tmp_name <- purrr::map_chr(i, ~paste0(., "_ggheatmap"))assign(tmp_name, tmp_ggheatmap)print(tmp_name)rm(list =ls(pattern ="tmp_"))} objects(pattern ="_ggheatmap")```## Autocorrelation Plots<br/>::: {.column-body}::: {.panel-tabset}#### Edaphic properties```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5edaphic_cor_ggheatmap```<small>`r caption_fig_its("its_autocor_edaph_plots")`</small>#### Edaphic properties (normalized)```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5edaphic_norm_cor_ggheatmap```<small>`r caption_fig_its("its_autocor_edaph_norm_plots")`</small>:::::: {.panel-tabset}#### Functional responses```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5soil_funct_cor_ggheatmap```<small>`r caption_fig_its("its_autocor_funct_plots")`</small>#### Functional responses (normalized)```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5soil_funct_norm_cor_ggheatmap```<small>`r caption_fig_its("its_autocor_funct_norm_plots")`</small>:::::: {.panel-tabset}#### Temperature adaptation ```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5temp_adapt_cor_ggheatmap```<small>`r caption_fig_its("its_autocor_temp_plots")`</small>#### Temperature adaptation (normalized)```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5temp_adapt_norm_cor_ggheatmap```<small>`r caption_fig_its("its_autocor_temp_norm_plots")`</small>::::::```{r}#| echo: falseauto_cor_figs <-ggarrange( edaphic_cor_ggheatmap, edaphic_norm_cor_ggheatmap, soil_funct_cor_ggheatmap, soil_funct_norm_cor_ggheatmap, temp_adapt_cor_ggheatmap, temp_adapt_norm_cor_ggheatmap,ncol =2, nrow =3, common.legend =FALSE)auto_cor_figsdev.off()png("files/metadata/figures/its18_auto_cor_figs.png",height =32, width =24, units ='cm', res =600, bg ="white")auto_cor_figsdev.off()pdf("files/metadata/figures/its18_auto_cor_figs.pdf",height =10, width =12)auto_cor_figsdev.off()```Now we can remove parameters based on the autocorrelation analysis:A) Environmental and edaphic properties: TEB, DON, Na, Al, Ca.B) Microbial functional responses: micN, micNP, enzCN, enzCP, BP_ase, CE_ase, LP_ase, N_ase, P_ase.C) Temperature adaptation properties: NUE, PUE, P_Q10, SI.```{r}#| echo: false#| eval: falseobjects(pattern ="_cor_ggheatmap")tmp_res <- soil_funct_norm_cor_ggheatmap$datavalue_choice <-0.7tmp_res %>%filter(value > value_choice | value <-value_choice) %>%filter_("Var1 != Var2")#tmp_res %>% filter(Var1 == "DOCN" | Var2 == "DOCN") %>% filter_("Var1 != Var2")write.table(tmp_res %>%filter(value > value_choice | value <-value_choice) %>%filter_("Var1 != Var2"), "soil_funct_norm.txt", sep ="\t", quote =FALSE, row.names =FALSE)``````{r}edaphic_remove <-c("TEB", "DON", "Na", "Al", "Ca")soil_funct_remove <-c("micN", "micNP", "enzCN", "enzCP", "BP_ase", "CE_ase", "LP_ase", "N_ase", "P_ase")temp_adapt_remove <-c("NUE", "PUE", "P_Q10", "SI")``````{r}#| code-fold: truetmp_df <- its18_select_mc_splittmp_df$edaphic <- tmp_df$edaphic[, !names(tmp_df$edaphic) %in% edaphic_remove]tmp_df$soil_funct <- tmp_df$soil_funct[, !names(tmp_df$soil_funct) %in% soil_funct_remove]tmp_df$temp_adapt <- tmp_df$temp_adapt[, !names(tmp_df$temp_adapt) %in% temp_adapt_remove]its18_select_mc_split_no_ac <- tmp_dfrm(list =ls(pattern ="tmp_"))tmp_df <- its18_select_mc_norm_splittmp_df$edaphic <- tmp_df$edaphic[, !names(tmp_df$edaphic) %in% edaphic_remove]tmp_df$soil_funct <- tmp_df$soil_funct[, !names(tmp_df$soil_funct) %in% soil_funct_remove]tmp_df$temp_adapt <- tmp_df$temp_adapt[, !names(tmp_df$temp_adapt) %in% temp_adapt_remove]its18_select_mc_norm_split_no_ac <- tmp_dfrm(list =ls(pattern ="tmp_"))```## Dissimilarity Correlation TestsLet's see if any on the metadata groups are significantly correlated with the community data. Basically, we create distance matrices for the community data and each metadata group and then run Mantel tests for all comparisons. For the community data we calculate Bray-Curtis distances for the community data and Euclidean distances for the metadata. We use the function `mantel.test` from the `ape` package and `mantel` from the `vegan` package for the analyses. In summary, we test both `mantel.test` and `mantel` on Bray-Curtis distance community distances against Euclidean distances for each metadata group (`edaphic`, `soil_funct`, `temp_adapt`) **a**) before normalizing and before removing autocorrelated parameters, **b**) before normalizing and after removing autocorrelated parameters, **c**) after normalizing and before removing autocorrelated parameters, and **d**) after normalizing and after removing autocorrelated parameters.```{r}#| code-fold: trueman_df <-c("its18_select_mc_split", "its18_select_mc_split_no_ac", "its18_select_mc_norm_split", "its18_select_mc_norm_split_no_ac")for (i in man_df) { tmp_get <-get(i) tmp_dm_otu <-as.matrix(vegdist(t(tmp_get$data_loaded), method ="bray", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE))# EDAPHIC tmp_dm_md_edaphic <-as.matrix(vegdist(tmp_get$edaphic[, 8:ncol(tmp_get$edaphic)], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man1_edaphic <-mantel.test(tmp_dm_otu, tmp_dm_md_edaphic, nperm =999, graph =FALSE, alternative ="two.sided") tmp_man2_edaphic <-mantel(tmp_dm_otu, tmp_dm_md_edaphic, permutations =999)# SOIL FUNCT tmp_dm_md_soil_funct <-as.matrix(vegdist(tmp_get$soil_funct[, 8:ncol(tmp_get$soil_funct)], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man1_soil_funct <-mantel.test(tmp_dm_otu, tmp_dm_md_soil_funct, nperm =999, graph =FALSE, alternative ="two.sided") tmp_man2_soil_funct <-mantel(tmp_dm_otu, tmp_dm_md_soil_funct, permutations =999)# TEMP ADAPT tmp_dm_md_temp_adapt <-as.matrix(vegdist(tmp_get$temp_adapt[, 8:ncol(tmp_get$temp_adapt)], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man1_temp_adapt <-mantel.test(tmp_dm_otu, tmp_dm_md_temp_adapt, nperm =999, graph =FALSE, alternative ="two.sided") tmp_man2_temp_adapt <-mantel(tmp_dm_otu, tmp_dm_md_temp_adapt, permutations =999) tmp_name <- purrr::map_chr(i, ~paste0(., "_mantel_tests")) tmp_df <-list(edaphic_ape_man = tmp_man1_edaphic, edaphic_vegan_man = tmp_man2_edaphic,soil_funct_ape_man = tmp_man1_soil_funct, soil_funct_vegan_man = tmp_man2_soil_funct,temp_adapt_ape_man = tmp_man1_temp_adapt, temp_adapt_vegan_man = tmp_man2_temp_adapt)assign(tmp_name, tmp_df)print(tmp_name)rm(list =ls(pattern ="tmp_"))}``````{r}#| echo: falsetmp_objects <-c("its18_select_mc_split_mantel_tests", "its18_select_mc_split_no_ac_mantel_tests","its18_select_mc_norm_split_mantel_tests","its18_select_mc_norm_split_no_ac_mantel_tests")tmp_norm <-data.frame(c("no", "no", "yes", "yes"))tmp_ac <-data.frame(c("no", "yes", "no", "yes"))its18_ps_pime_mantel_summary <- dplyr::bind_cols(tmp_norm, tmp_ac) %>% dplyr::rename("normalized?"=1) %>% dplyr::rename("AC removed?"=2)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$edaphic_ape_man$p,get(tmp_objects[2])$edaphic_ape_man$p,get(tmp_objects[3])$edaphic_ape_man$p,get(tmp_objects[4])$edaphic_ape_man$p))its18_ps_pime_mantel_summary <- dplyr::bind_cols(its18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("edaphic_ape"=3)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$edaphic_vegan_man$signif,get(tmp_objects[2])$edaphic_vegan_man$signif,get(tmp_objects[3])$edaphic_vegan_man$signif,get(tmp_objects[4])$edaphic_vegan_man$signif))its18_ps_pime_mantel_summary <- dplyr::bind_cols(its18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("edaphic_vegan"=4)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$soil_funct_ape_man$p,get(tmp_objects[2])$soil_funct_ape_man$p,get(tmp_objects[3])$soil_funct_ape_man$p,get(tmp_objects[4])$soil_funct_ape_man$p))its18_ps_pime_mantel_summary <- dplyr::bind_cols(its18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("soil_funct_ape"=5)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$soil_funct_vegan_man$signif,get(tmp_objects[2])$soil_funct_vegan_man$signif,get(tmp_objects[3])$soil_funct_vegan_man$signif,get(tmp_objects[4])$soil_funct_vegan_man$signif))its18_ps_pime_mantel_summary <- dplyr::bind_cols(its18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("soil_funct_vegan"=6)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$temp_adapt_ape_man$p,get(tmp_objects[2])$temp_adapt_ape_man$p,get(tmp_objects[3])$temp_adapt_ape_man$p,get(tmp_objects[4])$temp_adapt_ape_man$p))its18_ps_pime_mantel_summary <- dplyr::bind_cols(its18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("temp_adapt_ape"=7)tmp_pvalue <-data.frame(c(get(tmp_objects[1])$temp_adapt_vegan_man$signif,get(tmp_objects[2])$temp_adapt_vegan_man$signif,get(tmp_objects[3])$temp_adapt_vegan_man$signif,get(tmp_objects[4])$temp_adapt_vegan_man$signif))its18_ps_pime_mantel_summary <- dplyr::bind_cols(its18_ps_pime_mantel_summary, tmp_pvalue) %>% dplyr::rename("temp_adapt_vegan"=8)rm(list =ls(pattern ="tmp_"))```### Dissimilarity Correlation Results<small>`r caption_tab_its("its_mantel_summ")`</small>```{r}#| echo: false#| eval: trueseq_table <- its18_ps_pime_mantel_summaryreactable(seq_table,defaultColDef =colDef(header =function(value) gsub("_", " ", value, fixed =TRUE),cell =function(value) format(value, nsmall =3),align ="center", filterable =FALSE, sortable =FALSE, resizable =TRUE, minWidth =50,style =function(value) {if (value >0.05) { color <-"#2271B2"#green } elseif (value <0.05) { color <-"#B22271"#RED "#", "#" } else { color <-"#777" }list(color = color)} ), columns =list(`normalized?`=colDef(name ="normalized?", style =list(borderRight ="5px solid #eee"),headerStyle =list(borderRight ="5px solid #eee"), align ="left",minWidth =70 ),`AC removed?`=colDef(name ="AC removed?", align ="left",minWidth =70,headerStyle =list(borderRight ="5px solid #eee"), style =list(borderRight ="5px solid #eee") ),edaphic_ape =colDef(name ="ape", ),edaphic_vegan =colDef(name ="vegan" ),soil_funct_ape =colDef(name ="ape" ),soil_funct_vegan =colDef(name ="vegan" ),temp_adapt_ape =colDef(name ="ape" ),temp_adapt_vegan =colDef(name ="vegan" ) ), columnGroups =list(colGroup(name ="Environmental & edaphic", columns =c("edaphic_ape", "edaphic_vegan"), headerStyle =list(fontSize ="1.2em" ), ),colGroup(name ="Functional responses", columns =c("soil_funct_ape", "soil_funct_vegan"), headerStyle =list(fontSize ="1.2em" ) ),colGroup(name ="Temperature adaptation", columns =c("temp_adapt_ape", "temp_adapt_vegan"), headerStyle =list(fontSize ="1.2em") ) ),searchable =FALSE, defaultPageSize =15, showPagination =FALSE,pageSizeOptions =c(5, 10, nrow(seq_table)), showPageSizeOptions =TRUE, highlight =TRUE, bordered =TRUE, striped =TRUE, compact =TRUE, wrap =FALSE, showSortable =FALSE, fullWidth =TRUE,theme =reactableTheme(style =list(fontSize ="0.8em")))rm(seq_table)```Moving on.## Best Subset of VariablesNow we want to know which of the metadata parameters are the most strongly correlated with the community data. For this we use the `bioenv` function from the `vegan` package. `bioenv`---*Best Subset of Environmental Variables with Maximum (Rank) Correlation with Community Dissimilarities*---finds the best subset of environmental variables, so that the Euclidean distances of scaled environmental variables have the maximum (rank) correlation with community dissimilarities. Since we know that each of the Mantel tests we ran above are significant, here we will use the metadata set where autocorrelated parameters were removed and the remainder of the parameters were normalized (where applicable based on the Shapiro tests). We run `bioenv` against the three groups of metadata parameters. We then run `bioenv` again, but this time against the individual parameters identified as significantly correlated.### Edaphic Properties```{r}#| code-fold: truetmp_comm <-data.frame(t(its18_select_mc_norm_split_no_ac$data_loaded))tmp_env <-data.frame(its18_select_mc_norm_split_no_ac$edaphic)tmp_env[,1:8] <-NULLedaphic_bioenv <-bioenv(wisconsin(tmp_comm), tmp_env, method ="spearman", index ="bray", upto =ncol(tmp_env), metric ="euclidean")bioenv_list <- edaphic_bioenv$models[[edaphic_bioenv$whichbest]]$bestbioenv_best <-bioenvdist(edaphic_bioenv, which ="best")for (i in bioenv_list) { tmp_dp <-data.frame(edaphic_bioenv$x) tmp_md <-as.matrix(vegdist(tmp_dp[[i]], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man <-mantel(bioenv_best, tmp_md, permutations =999, method ="spearman") tmp_md_name <-names(tmp_dp)[[i]] tmp_name <- purrr::map_chr(tmp_md_name, ~paste0(., "_bioenv_mantel_test"))assign(tmp_name, tmp_man)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bioenv_mantel_test")edaphic_bioenv_ind_mantel <-list(AST = AST_bioenv_mantel_test)rm(list =ls(pattern ="_bioenv_mantel_test"))``````{r}#| echo: false#| eval: trueedaphic_bioenv```<details><summary>Show the results of individual edaphic metadata Mantel tests</summary>```{r}#| echo: false#| eval: trueedaphic_bioenv_ind_mantel```</details>`bioenv` found the following edaphic properties significantly correlated with the community data: **`r row.names(summary(edaphic_bioenv_ind_mantel))`**### Soil Functional Response```{r}#| code-fold: truetmp_comm <-data.frame(t(its18_select_mc_norm_split_no_ac$data_loaded))tmp_env <-data.frame(its18_select_mc_norm_split_no_ac$soil_funct)tmp_env[,1:8] <-NULLsoil_funct_bioenv <-bioenv(wisconsin(tmp_comm), tmp_env, method ="spearman", index ="bray", upto =ncol(tmp_env), metric ="euclidean")bioenv_list <- soil_funct_bioenv$models[[soil_funct_bioenv$whichbest]]$bestbioenv_best <-bioenvdist(soil_funct_bioenv, which ="best")for (i in bioenv_list) { tmp_dp <-data.frame(soil_funct_bioenv$x) tmp_md <-as.matrix(vegdist(tmp_dp[[i]], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man <-mantel(bioenv_best, tmp_md, permutations =999, method ="spearman") tmp_md_name <-names(tmp_dp)[[i]] tmp_name <- purrr::map_chr(tmp_md_name, ~paste0(., "_bioenv_mantel_test"))assign(tmp_name, tmp_man)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bioenv_mantel_test")soil_funct_bioenv_ind_mantel <-list(enzNP = enzNP_bioenv_mantel_test, PX_ase = PX_ase_bioenv_mantel_test,XY_ase = XY_ase_bioenv_mantel_test)rm(list =ls(pattern ="_bioenv_mantel_test"))``````{r}#| echo: false#| eval: truesoil_funct_bioenv```<details><summary>Show the results of individual functional response metadata Mantel tests</summary>```{r}#| echo: false#| eval: truesoil_funct_bioenv_ind_mantel```</details>`bioenv` found the following soil functions significantly correlated with the community data: **`r row.names(summary(soil_funct_bioenv_ind_mantel))`**### Temperature Adaptation```{r}#| code-fold: truetmp_comm <-data.frame(t(its18_select_mc_norm_split_no_ac$data_loaded))tmp_env <-data.frame(its18_select_mc_norm_split_no_ac$temp_adapt)tmp_env[,1:8] <-NULLtemp_adapt_bioenv <-bioenv(wisconsin(tmp_comm), tmp_env, method ="spearman", index ="bray", upto =ncol(tmp_env), metric ="euclidean")bioenv_list <- temp_adapt_bioenv$models[[temp_adapt_bioenv$whichbest]]$bestbioenv_best <-bioenvdist(temp_adapt_bioenv, which ="best")for (i in bioenv_list) { tmp_dp <-data.frame(temp_adapt_bioenv$x) tmp_md <-as.matrix(vegdist(tmp_dp[[i]], method ="euclidean", binary =FALSE, diag =TRUE, upper =TRUE, na.rm =FALSE)) tmp_man <-mantel(bioenv_best, tmp_md, permutations =999, method ="spearman") tmp_md_name <-names(tmp_dp)[[i]] tmp_name <- purrr::map_chr(tmp_md_name, ~paste0(., "_bioenv_mantel_test"))assign(tmp_name, tmp_man)rm(list =ls(pattern ="tmp_"))}objects(pattern ="_bioenv_mantel_test")temp_adapt_bioenv_ind_mantel <-list(XY_Q10 = XY_Q10_bioenv_mantel_test,Tmin = Tmin_bioenv_mantel_test)rm(list =ls(pattern ="_bioenv_mantel_test"))``````{r}#| echo: false#| eval: truetemp_adapt_bioenv```<details><summary>Show the results of individual temperature adaptation metadata Mantel tests</summary>```{r}#| echo: false#| eval: truetemp_adapt_bioenv_ind_mantel```</details>`bioenv` found the following temperature adaptations significantly correlated with the community data: **`r row.names(summary(temp_adapt_bioenv_ind_mantel))`**## Distance-based RedundancyNow we turn our attention to distance-based redundancy analysis (dbRDA), an ordination method similar to Redundancy Analysis (rda) but it allows non-Euclidean dissimilarity indices, such as Manhattan or Bray–Curtis distance. For this, we use `capscale` from the `vegan` package. `capscale` is a constrained versions of metric scaling (principal coordinates analysis), which are based on the Euclidean distance but can be used, and are more useful, with other dissimilarity measures. The functions can also perform unconstrained principal coordinates analysis, optionally using extended dissimilarities.For each of the three metadata subsets, we perform the following steps:1) Run `rankindex` to compare metadata and community dissimilarity indices for gradient detection. This will help us select the best dissimilarity metric to use.2) Run `capscale` for distance-based redundancy analysis.3) Run `envfit` to fit environmental parameters onto the ordination. This function basically calculates correlation scores between the metadata parameters and the ordination axes. 4) Select metadata parameters significant for `bioenv` (see above) and/or `envfit` analyses.5) Run `envfit` on ASVs.6) Plot the ordination and vector overlays. ### Edaphic Properties```{r}#| code-fold: truetmp_md <- its18_select_mc_norm_split_no_ac$edaphictmp_md$TREAT_T <-as.character(tmp_md$TREAT_T)tmp_comm <-data.frame(t(its18_select_mc_norm_split_no_ac$data_loaded))edaphic_rank <-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow","bra", "kul"), stepacross =FALSE, method ="spearman")``````{r}#| echo: false#| eval: trueedaphic_rank```Let's run `capscale` using Bray-Curtis. Note, we have `r ncol(its18_select_mc_norm_split_no_ac$edaphic) - 8` metadata parameters in this group but, for some reason, `capscale` only works with 13 parameters. This may have to do with degrees of freedom? * Starting properties: AST, H2O, N, P, Al, Ca, Fe, K, Mg, Mn, Na, TEB, ECEC, pH, NH4, NO3, PO4, DOC, DON, DOCN.* Autocorrelated removed: TEB, DON, Na, Al, Ca.* Remove for capscale: Mg, Mn, Na, Al, Fe, K```{r}#| echo: false# All edaphic: AST, H2O, N, P, Al, Ca, Fe, K, Mg, Mn, Na, TEB, ECEC, pH, NH4, NO3, PO4, DOC, DON, DOCN# REMOVED edaphic: TEB, DON, Na, Al, Ca# Remaining edaphic: AST, H2O, N, P, Fe, K, Mg, Mn, ECEC, pH, NH4, NO3, PO4, DOC, DOCN# only works with 11# Removed Mg + Mn + Na + Al + Fe + K# TEST with , metaMDSdist = TRUE AND/OR , metaMDS = TRUE``````{r}edaphic_cap <-capscale(tmp_comm ~ AST + H2O + N + P + ECEC + pH + NH4 + NO3 + PO4 + DOC + DOCN, tmp_md, dist ="bray")colnames(tmp_md)``````{r}#| echo: false#| eval: trueedaphic_cap```Now we can look at the variance against each principal component. ```{r}#| echo: false#| eval: truestats::screeplot(edaphic_cap)```And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The `ggplot` function `autoplot` stores these data in a more accessible way than the raw results from `capscale````{r}#| eval: truebase::plot(edaphic_cap) tmp_auto_plt <- ggplot2::autoplot(edaphic_cap, arrows =TRUE)tmp_auto_plt``````{r}#| echo: falseanova(edaphic_cap) # overall test of the significant of the analysisanova(edaphic_cap, by ="axis", perm.max =500) # test axes for significanceanova(edaphic_cap, by ="terms", permu =500) # test for sign. environ. variables```Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function `scores` which gets species or site scores from the ordination.```{r}#| code-fold: truetmp_samp_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="sites")tmp_samp_scores[,1] <-NULLtmp_samp_scores <- tmp_samp_scores %>% dplyr::rename(SampleID = Label)tmp_md_sub <- tmp_md[, 1:4]tmp_md_sub <- tmp_md_sub %>% tibble::rownames_to_column("SampleID")edaphic_plot_data <- dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")```Now we have a new data frame that contains sample details and capscale values. ```{r}#| echo: false#| eval: trueedaphic_plot_data```We can then do the same with the metadata vectors. Here though we only need the scores and parameter name. ```{r}#| code-fold: trueedaphic_md_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="biplot")edaphic_md_scores[,1] <-NULLedaphic_md_scores <- edaphic_md_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")``````{r}#| echo: false#| eval: trueedaphic_md_scores```Let's run some quick correlations of metadata with ordination axes to see which parameters are significant. For this we use the vegan function `envfit`.```{r}#| code-fold: truetmp_samp_scores_sub <- edaphic_plot_data[, 6:7]tmp_samp_scores_sub <-as.matrix(tmp_samp_scores_sub)tmp_param_list <- edaphic_md_scores$parameterstmp_md_sub <-subset(tmp_md, select = tmp_param_list)envfit_edaphic_md <-envfit(tmp_samp_scores_sub, tmp_md_sub,perm =1000, choices =c(1, 2))``````{r}#| echo: false#| eval: trueenvfit_edaphic_md``````{r}#| code-fold: trueedaphic_md_signif_hits <- base::subset(envfit_edaphic_md$vectors$pvals, c(envfit_edaphic_md$vectors$pvals <0.05& envfit_edaphic_md$vectors$r >0.4))edaphic_md_signif_hits <-data.frame(edaphic_md_signif_hits)edaphic_md_signif_hits <-rownames(edaphic_md_signif_hits)edaphic_md_signif <- edaphic_md_scores[edaphic_md_scores$parameters %in% edaphic_md_signif_hits,]edaphic_md_signif$parameters````envfit` found that `r edaphic_md_signif$parameters` were significantly correlated. Now let's see if the same parameters are significant for the `envfit` and `bioenv` analyses.```{r}#| echo: false#| eval: true#| results: hold#| comment: ''print("Significant parameters from bioenv analysis.")row.names(summary(edaphic_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")edaphic_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(edaphic_bioenv_ind_mantel)), edaphic_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(edaphic_md_signif$parameters, row.names(summary(edaphic_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")edaphic_sig_diff <- base::union(edaphic_md_signif$parameters, row.names(summary(edaphic_bioenv_ind_mantel)))edaphic_sig_diff``````{r}#| code-fold: truenew_edaphic_md_signif_hits <- edaphic_sig_diff#new_edaphic_md_signif_hits <- append(edaphic_md_signif_hits, edaphic_sig_diff)edaphic_md_signif_all <- edaphic_md_scores[edaphic_md_scores$parameters %in% new_edaphic_md_signif_hits,]```Check. Next, we run `envfit` for the ASVs.```{r}#| code-fold: trueenvfit_edaphic_asv <-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10],perm =1000, choices =c(1, 2))edaphic_asv_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="species")edaphic_asv_scores <- edaphic_asv_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")edaphic_asv_scores[,1] <-NULL``````{r}#| echo: false#| eval: trueenvfit_edaphic_asv``````{r}#| code-fold: trueedaphic_asv_signif_hits <- base::subset(envfit_edaphic_asv$vectors$pvals, c(envfit_edaphic_asv$vectors$pvals <0.05& envfit_edaphic_asv$vectors$r >0.5))edaphic_asv_signif_hits <-data.frame(edaphic_asv_signif_hits)edaphic_asv_signif_hits <-rownames(edaphic_asv_signif_hits)edaphic_asv_signif <- edaphic_asv_scores[edaphic_asv_scores$parameters %in% edaphic_asv_signif_hits,]``````{r}#| echo: false#| eval: trueedaphic_asv_signif``````{r}#| code-fold: trueedaphic_md_signif_all$variable_type <-"metadata"edaphic_asv_signif$variable_type <-"ASV"edaphic_bioplot_data <-rbind(edaphic_md_signif_all, edaphic_asv_signif)```The last thing to do is categorize parameters scores and ASV scores into different variable types for plotting.```{r}#| code-fold: trueedaphic_bioplot_data_md <-subset(edaphic_bioplot_data, edaphic_bioplot_data$variable_type =="metadata")edaphic_bioplot_data_asv <-subset(edaphic_bioplot_data, edaphic_bioplot_data$variable_type =="ASV")```<details><summary>Show the code for the plot</summary>```{r}#| echo: falseedaphic_cap_vals <-data.frame(edaphic_cap$CCA$eig[1:2])edaphic_cap1 <-signif((edaphic_cap_vals[1,] *100), digits=3)edaphic_cap2 <-signif((edaphic_cap_vals[2,] *100), digits=3)cpa1_lab <-paste("CAP1", " (", edaphic_cap1, "%)", sep ="")cpa2_lab <-paste("CAP2", " (", edaphic_cap2, "%)", sep ="")swel_col <-c("#2271B2", "#71B222", "#B22271")edaphic_plot <-ggplot(edaphic_plot_data) +geom_point(mapping =aes(x = CAP1, y = CAP2, shape = TREAT,colour = TREAT_T), size =4) +scale_colour_manual(values = swel_col) +# geom_text(data = edaphic_plot_data, aes(x = CAP1, y = CAP2, #UNCOMMENT to add sample labels# label = SampleID), size = 3) + geom_segment(aes(x =0, y =0, xend = CAP1, yend = CAP2),data = edaphic_bioplot_data_md, linetype ="solid",arrow =arrow(length =unit(0.3, "cm")), size =0.4, color ="#191919", inherit.aes =FALSE) +geom_text(data = edaphic_bioplot_data_md, aes(x = CAP1, y = CAP2, label = parameters), size =3, nudge_x =0.1, nudge_y =0.05) +## USe this code to overlay ASV vectors#geom_segment(aes(x = 0, y = 0, xend = CAP1, yend = CAP2),# data = edaphic_bioplot_data_asv, linetype = "solid",# arrow = arrow(length = unit(0.3, "cm")), size = 0.2, # color = "#676767") +#geom_text(data = edaphic_bioplot_data_asv, # aes(x = CAP1, y = CAP2, label = parameters), size = 2.5,# nudge_x = 0.1, nudge_y = 0.05) +theme_classic(base_size =12) +labs(title ="Capscale Analysis",subtitle ="Edaphic properties", x = cpa1_lab, y = cpa2_lab)edaphic_plot <- edaphic_plot +coord_fixed() +theme(aspect.ratio=1)edaphic_plotpng("files/metadata/figures/its18_edaphic_capscale.png",height =16, width =20, units ='cm', res =600, bg ="white")edaphic_plotinvisible(dev.off())pdf("files/metadata/figures/its18_edaphic_capscale.pdf",height =5, width =6)edaphic_plotdev.off()```</details>```{r}#| echo: falserm(list =ls(pattern ="tmp_"))```### Soil Functional Response```{r}#| code-fold: truetmp_md <- its18_select_mc_norm_split_no_ac$soil_functtmp_md$TREAT_T <-as.character(tmp_md$TREAT_T)tmp_comm <-data.frame(t(its18_select_mc_norm_split_no_ac$data_loaded))soil_funct_rank <-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow", "bra", "kul"), stepacross =FALSE, method ="spearman")``````{r}#| echo: false#| eval: truesoil_funct_rank```Let's run `capscale` using Bray-Curtis Note, we have `r ncol(its18_select_mc_norm_split_no_ac$soil_funct) - 8` metadata parameters in this group but, for some reason, `capscale` only works with 13 parameters. This may have to do with degrees of freedom? * Starting properties: micC, micN, micP, micCN, micCP, micNP, AG_ase, BG_ase, BP_ase, CE_ase, P_ase, N_ase, S_ase, XY_ase, LP_ase, PX_ase, CO2, enzCN, enzCP, enzNP* Autocorrelated removed: micN, micNP, enzCN, enzCP, BP_ase, CE_ase, LP_ase, N_ase, P_ase* Remove for capscale: NONE```{r}#| echo: false# All soil prop: micC, micN, micP, micCN, micCP, micNP, AG_ase, BG_ase, BP_ase, CE_ase, P_ase, N_ase, S_ase, XY_ase, LP_ase, PX_ase, CO2, enzCN, enzCP, enzNP# REMOVED soil prop: micN, micNP, enzCN, enzCP, BP_ase, CE_ase, LP_ase, N_ase, P_ase# Remaining soil prop: micC, micP, micCN, micCP, AG_ase, BG_ase, S_ase, XY_ase, PX_ase, CO2, enzNP# only works with 11# capscale Removed NONE``````{r}soil_funct_cap <-capscale(tmp_comm ~ micC + micP + micCN + micCP + AG_ase + BG_ase + S_ase + XY_ase + PX_ase + CO2 + enzNP, tmp_md, dist ="bray")``````{r}#| echo: false#| eval: truesoil_funct_cap```Now we can look at the variance against each principal component. ```{r}#| echo: false#| eval: truescreeplot(soil_funct_cap)```And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The `ggplot` function `autoplot` stores these data in a more accessible way than the raw results from `capscale````{r}#| eval: truebase::plot(soil_funct_cap) tmp_auto_plt <-autoplot(soil_funct_cap, arrows =TRUE)tmp_auto_plt``````{r}#| echo: falseanova(soil_funct_cap) # overall test of the significant of the analysisanova(soil_funct_cap, by ="axis", perm.max=500) # test axes for significanceanova(soil_funct_cap, by ="terms", permu=500) # test for sign. environ. variables```Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function `scores` which gets species or site scores from the ordination.```{r}#| code-fold: truetmp_samp_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="sites")tmp_samp_scores[,1] <-NULLtmp_samp_scores <- tmp_samp_scores %>% dplyr::rename(SampleID = Label)tmp_md_sub <- tmp_md[, 1:4]tmp_md_sub <- tmp_md_sub %>% tibble::rownames_to_column("SampleID")soil_funct_plot_data <- dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")```Now we have a new data frame that contains sample details and capscale values. ```{r}#| echo: false#| eval: truesoil_funct_plot_data```We can then do the same with the metadata vectors. Here though we only need the scores and parameter name. ```{r}#| code-fold: truesoil_funct_md_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="biplot")soil_funct_md_scores[,1] <-NULLsoil_funct_md_scores <- soil_funct_md_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")``````{r}#| echo: false#| eval: truesoil_funct_md_scores```Let's run some quick correlations of metadata with ordination axes to see which parameters are significant. For this we use the vegan function `envfit`.```{r}#| code-fold: truetmp_samp_scores_sub <- soil_funct_plot_data[, 6:7]tmp_samp_scores_sub <-as.matrix(tmp_samp_scores_sub)tmp_param_list <- soil_funct_md_scores$parameterstmp_md_sub <-subset(tmp_md, select = tmp_param_list)envfit_soil_funct_md <-envfit(tmp_samp_scores_sub, tmp_md_sub,perm =1000, choices =c(1, 2))``````{r}#| echo: false#| eval: trueenvfit_soil_funct_md``````{r}#| code-fold: truesoil_funct_md_signif_hits <- base::subset(envfit_soil_funct_md$vectors$pvals, c(envfit_soil_funct_md$vectors$pvals <0.05& envfit_soil_funct_md$vectors$r >0.4))soil_funct_md_signif_hits <-data.frame(soil_funct_md_signif_hits)soil_funct_md_signif_hits <-rownames(soil_funct_md_signif_hits)soil_funct_md_signif <- soil_funct_md_scores[soil_funct_md_scores$parameters %in% soil_funct_md_signif_hits,]soil_funct_md_signif$parameters````envfit` found that `r soil_funct_md_signif$parameters` were significantly correlated. Now let's see if the same parameters are significant for the `envfit` and `bioenv` analyses.```{r}#| echo: false#| eval: true#| results: hold#| comment: '' print("Significant parameters from bioenv analysis.")row.names(summary(soil_funct_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")soil_funct_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(soil_funct_bioenv_ind_mantel)), soil_funct_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(soil_funct_md_signif$parameters, row.names(summary(soil_funct_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")soil_funct_sig_diff <- base::union(soil_funct_md_signif$parameters, row.names(summary(soil_funct_bioenv_ind_mantel)))soil_funct_sig_diff``````{r}#| code-fold: true#new_soil_funct_md_signif_hits <- append(soil_funct_md_signif_hits, soil_funct_sig_diff)new_soil_funct_md_signif_hits <- soil_funct_sig_diffsoil_funct_md_signif_all <- soil_funct_md_scores[soil_funct_md_scores$parameters %in% new_soil_funct_md_signif_hits,]```Check. Next, we run `envfit` for the ASVs.```{r}#| code-fold: trueenvfit_soil_funct_asv <-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10], perm =1000, choices =c(1, 2))soil_funct_asv_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="species")soil_funct_asv_scores <- soil_funct_asv_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")soil_funct_asv_scores[,1] <-NULL``````{r}#| echo: false#| eval: trueenvfit_soil_funct_asv``````{r}#| code-fold: truesoil_funct_asv_signif_hits <- base::subset(envfit_soil_funct_asv$vectors$pvals, c(envfit_soil_funct_asv$vectors$pvals <0.05& envfit_soil_funct_asv$vectors$r >0.5))soil_funct_asv_signif_hits <-data.frame(soil_funct_asv_signif_hits)soil_funct_asv_signif_hits <-rownames(soil_funct_asv_signif_hits)soil_funct_asv_signif <- soil_funct_asv_scores[soil_funct_asv_scores$parameters %in% soil_funct_asv_signif_hits,]``````{r}#| echo: false#| eval: truesoil_funct_asv_signif``````{r}#| code-fold: truesoil_funct_md_signif_all$variable_type <-"metadata"soil_funct_asv_signif$variable_type <-"ASV"soil_funct_bioplot_data <-rbind(soil_funct_md_signif_all, soil_funct_asv_signif)```The last thing to do is categorize parameters scores and ASV scores into different variable types for plotting.```{r}#| code-fold: truesoil_funct_bioplot_data_md <-subset(soil_funct_bioplot_data, soil_funct_bioplot_data$variable_type =="metadata")soil_funct_bioplot_data_asv <-subset(soil_funct_bioplot_data, soil_funct_bioplot_data$variable_type =="ASV")```<details><summary>Show the code for the plot</summary>```{r}soil_funct_cap_vals <-data.frame(soil_funct_cap$CCA$eig[1:2])soil_funct_cap1 <-signif((soil_funct_cap_vals[1,] *100), digits=3)soil_funct_cap2 <-signif((soil_funct_cap_vals[2,] *100), digits=3)cpa1_lab <-paste("CAP1", " (", soil_funct_cap1, "%)", sep ="")cpa2_lab <-paste("CAP2", " (", soil_funct_cap2, "%)", sep ="")swel_col <-c("#2271B2", "#71B222", "#B22271")soil_funct_plot <-ggplot(soil_funct_plot_data) +geom_point(mapping =aes(x = CAP1, y = CAP2, shape = TREAT,colour = TREAT_T), size =4) +scale_colour_manual(values = swel_col) +# geom_text(data = soil_funct_plot_data, aes(x = CAP1, y = CAP2, #UNCOMMENT to add sample labels# label = SampleID), size = 3) + geom_segment(aes(x =0, y =0, xend = CAP1, yend = CAP2),data = soil_funct_bioplot_data_md, linetype ="solid",arrow =arrow(length =unit(0.3, "cm")), size =0.4, color ="#191919") +geom_text(data = soil_funct_bioplot_data_md, aes(x = CAP1, y = CAP2, label = parameters), size =3,nudge_x =0.1, nudge_y =0.05) +## USE this code to overlay ASV vestors#geom_segment(aes(x = 0, y = 0, xend = CAP1, yend = CAP2),# data = soil_funct_bioplot_data_asv, linetype = "solid",# arrow = arrow(length = unit(0.3, "cm")), size = 0.2,# color = "#676767") +#geom_text(data = soil_funct_bioplot_data_asv, # aes(x = CAP1, y = CAP2, label = parameters), size = 2.5,# nudge_x = 0.05, nudge_y = 0.05) +theme_classic(base_size =12) +labs(title ="Capscale Analysis",subtitle ="Soil Functional Response", x = cpa1_lab, y = cpa2_lab)soil_funct_plot <- soil_funct_plot +coord_fixed() +theme(aspect.ratio=1)soil_funct_plotpng("files/metadata/figures/its18_soil_funct_capscale.png",height =16, width =20, units ='cm', res =600, bg ="white")soil_funct_plotinvisible(dev.off())pdf("files/metadata/figures/its18_soil_funct_capscale.pdf",height =5, width =6)soil_funct_plotdev.off()```</details>```{r}#| echo: falserm(list =ls(pattern ="tmp_"))```### Temperature Adaptation```{r}#| code-fold: truetmp_md <- its18_select_mc_norm_split_no_ac$temp_adapttmp_md$TREAT_T <-as.character(tmp_md$TREAT_T)tmp_comm <-data.frame(t(its18_select_mc_norm_split_no_ac$data_loaded))temp_adapt_rank <-rankindex(tmp_md[, 8:ncol(tmp_md)], tmp_comm, indices =c("euc", "man", "gow", "bra", "kul"), stepacross =FALSE, method ="spearman")``````{r}#| echo: false#| eval: truetemp_adapt_rank```Let's run `capscale` using Bray-Curtis. Note, we have `r ncol(its18_select_mc_norm_split_no_ac$temp_adapt) - 8` metadata parameters in this group but, for some reason, `capscale` only works with 13 parameters. This may have to do with degrees of freedom? * Starting properties: AG_Q10, BG_Q10, BP_Q10, CE_Q10, P_Q10, N_Q10, S_Q10, XY_Q10, LP_Q10, PX_Q10, CUEcn, CUEcp, NUE, PUE, Tmin, SI* Autocorrelated removed: NUE, PUE, P_Q10, SI * Remove for capscale: S_Q10```{r}#| echo: false# All temp_adapt: AG_Q10, BG_Q10, BP_Q10, CE_Q10, P_Q10, N_Q10, S_Q10, XY_Q10, LP_Q10, PX_Q10, CUEcn, CUEcp, NUE, PUE, Tmin, SI# REMOVED edaphic: NUE, PUE, P_Q10, SI # Remaining edaphic: AG_Q10, BG_Q10, BP_Q10, CE_Q10, N_Q10, S_Q10, XY_Q10, LP_Q10, PX_Q10, CUEcn, CUEcp, Tmin# only works with 11# Removed: S_Q10``````{r}temp_adapt_cap <-capscale(tmp_comm ~ AG_Q10 + BG_Q10 + BP_Q10 + CE_Q10 + N_Q10 + XY_Q10 + LP_Q10 + PX_Q10 + CUEcn + CUEcp + Tmin, tmp_md, dist ="bray")``````{r}#| echo: false#| eval: truetemp_adapt_cap```Now we can look at the variance against each principal component. ```{r}#| echo: false#| eval: truescreeplot(temp_adapt_cap)```And then make some quick and dirty plots. This will also come in handy later when we need to parse out data a better plot visualization. The `ggplot` function `autoplot` stores these data in a more accessible way than the raw results from `capscale````{r}#| eval: truebase::plot(temp_adapt_cap) tmp_auto_plt <-autoplot(temp_adapt_cap, arrows =TRUE)tmp_auto_plt``````{r}#| echo: falseanova(temp_adapt_cap) # overall test of the significant of the analysisanova(temp_adapt_cap, by ="axis", perm.max =500) # test axes for significanceanova(temp_adapt_cap, by ="terms", permu =500) # test for sign. environ. variables```Next, we need to grab capscale scores for the samples and create a data frame of the first two dimensions. We will also need to add some of the sample details to the data frame. For this we use the vegan function `scores` which gets species or site scores from the ordination.```{r}#| code-fold: truetmp_samp_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="sites")tmp_samp_scores[,1] <-NULLtmp_samp_scores <- tmp_samp_scores %>% dplyr::rename(SampleID = Label)tmp_md_sub <- tmp_md[, 1:4]tmp_md_sub <- tmp_md_sub %>% tibble::rownames_to_column("SampleID")temp_adapt_plot_data <- dplyr::left_join(tmp_md_sub, tmp_samp_scores, by ="SampleID")```Now we have a new data frame that contains sample details and capscale values. ```{r}#| echo: false#| eval: truetemp_adapt_plot_data```We can then do the same with the metadata vectors. Here though we only need the scores and parameter name. ```{r}#| code-fold: truetemp_adapt_md_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="biplot")temp_adapt_md_scores[,1] <-NULLtemp_adapt_md_scores <- temp_adapt_md_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")``````{r}#| echo: false#| eval: truetemp_adapt_md_scores```Let's run some quick correlations of metadata with ordination axes to see which parameters are significant. For this we use the vegan function `envfit`.```{r}#| code-fold: truetmp_samp_scores_sub <- temp_adapt_plot_data[, 6:7]tmp_samp_scores_sub <-as.matrix(tmp_samp_scores_sub)tmp_param_list <- temp_adapt_md_scores$parameterstmp_md_sub <-subset(tmp_md, select = tmp_param_list)envfit_temp_adapt_md <-envfit(tmp_samp_scores_sub, tmp_md_sub,perm =1000, choices =c(1, 2))``````{r}#| echo: false#| eval: trueenvfit_temp_adapt_md``````{r}#| code-fold: truetemp_adapt_md_signif_hits <- base::subset(envfit_temp_adapt_md$vectors$pvals, c(envfit_temp_adapt_md$vectors$pvals <0.05& envfit_temp_adapt_md$vectors$r >0.4))temp_adapt_md_signif_hits <-data.frame(temp_adapt_md_signif_hits)temp_adapt_md_signif_hits <-rownames(temp_adapt_md_signif_hits)temp_adapt_md_signif <- temp_adapt_md_scores[temp_adapt_md_scores$parameters %in% temp_adapt_md_signif_hits,]````envfit` found that `r temp_adapt_md_signif$parameters` were significantly correlated. Now let's see if the same parameters are significant for the `envfit` and `bioenv` analyses.```{r}#| echo: false#| eval: true#| results: hold#| comment: '' print("Significant parameters from bioenv analysis.")row.names(summary(temp_adapt_bioenv_ind_mantel))cat("_____________________________________")cat("\n")print("Significant parameters from envfit analysis.")temp_adapt_md_signif$parameterscat("_____________________________________")cat("\n")print("Found in bioenv but not envfit.")base::setdiff(row.names(summary(temp_adapt_bioenv_ind_mantel)), temp_adapt_md_signif$parameters)cat("_____________________________________")cat("\n")print("Found in envfit but not bioenv.")base::setdiff(temp_adapt_md_signif$parameters, row.names(summary(temp_adapt_bioenv_ind_mantel)))cat("_____________________________________")cat("\n")print("Found in envfit and bioenv.")temp_adapt_sig_diff <- base::union(temp_adapt_md_signif$parameters, row.names(summary(temp_adapt_bioenv_ind_mantel)))temp_adapt_sig_diff``````{r}#| code-fold: true#new_temp_adapt_md_signif_hits <- base::append(temp_adapt_md_signif_hits, temp_adapt_sig_diff)new_temp_adapt_md_signif_hits <- temp_adapt_sig_diff[1:4]temp_adapt_md_signif_all <- temp_adapt_md_scores[temp_adapt_md_scores$parameters %in% new_temp_adapt_md_signif_hits,]```Check. Next, we run `envfit` for the ASVs.```{r}#| code-fold: trueenvfit_temp_adapt_asv <-envfit(tmp_samp_scores_sub, tmp_comm[, order(colSums(-tmp_comm))][, 1:10],perm =1000, choices =c(1, 2))temp_adapt_asv_scores <- dplyr::filter(tmp_auto_plt$plot_env$obj, Score =="species")temp_adapt_asv_scores <- temp_adapt_asv_scores %>% dplyr::mutate(parameters = Label, .before = CAP1) %>% tibble::column_to_rownames("Label")temp_adapt_asv_scores[,1] <-NULL``````{r}#| echo: false#| eval: trueenvfit_temp_adapt_asv``````{r}#| code-fold: truetemp_adapt_asv_signif_hits <- base::subset(envfit_temp_adapt_asv$vectors$pvals, c(envfit_temp_adapt_asv$vectors$pvals <0.05& envfit_temp_adapt_asv$vectors$r >0.5))temp_adapt_asv_signif_hits <-data.frame(temp_adapt_asv_signif_hits)temp_adapt_asv_signif_hits <-rownames(temp_adapt_asv_signif_hits)temp_adapt_asv_signif <- temp_adapt_asv_scores[temp_adapt_asv_scores$parameters %in% temp_adapt_asv_signif_hits,]``````{r}#| echo: false#| eval: truetemp_adapt_asv_signif``````{r}#| code-fold: truetemp_adapt_md_signif_all$variable_type <-"metadata"temp_adapt_asv_signif$variable_type <-"ASV"temp_adapt_bioplot_data <-rbind(temp_adapt_md_signif_all, temp_adapt_asv_signif)```The last thing to do is categorize parameters scores and ASV scores into different variable types for plotting.```{r}#| code-fold: truetemp_adapt_bioplot_data_md <-subset(temp_adapt_bioplot_data, temp_adapt_bioplot_data$variable_type =="metadata")temp_adapt_bioplot_data_asv <-subset(temp_adapt_bioplot_data, temp_adapt_bioplot_data$variable_type =="ASV")```<details><summary>Show the code for the plot</summary>```{r}temp_adapt_cap_vals <-data.frame(temp_adapt_cap$CCA$eig[1:2])temp_adapt_cap1 <-signif((temp_adapt_cap_vals[1,] *100), digits=3)temp_adapt_cap2 <-signif((temp_adapt_cap_vals[2,] *100), digits=3)cpa1_lab <-paste("CAP1", " (", temp_adapt_cap1, "%)", sep ="")cpa2_lab <-paste("CAP2", " (", temp_adapt_cap2, "%)", sep ="")swel_col <-c("#2271B2", "#71B222", "#B22271")temp_adapt_plot <-ggplot(temp_adapt_plot_data) +geom_point(mapping =aes(x = CAP1, y = CAP2, shape = TREAT,colour = TREAT_T), size =4) +scale_colour_manual(values = swel_col) +# geom_text(data = temp_adapt_plot_data, aes(x = CAP1, y = CAP2, #UNCOMMENT to add sample labels# label = SampleID), size = 3) + geom_segment(aes(x =0, y =0, xend = CAP1, yend = CAP2),data = temp_adapt_bioplot_data_md, linetype ="solid",arrow =arrow(length =unit(0.3, "cm")), size =0.4,color ="#191919", inherit.aes =FALSE) +geom_text(data = temp_adapt_bioplot_data_md, aes(x = CAP1, y = CAP2, label = parameters), size =3,nudge_x =0.1, nudge_y =0.05) +# uSe to include ASV vectors#geom_segment(aes(x = 0, y = 0, xend = CAP1, yend = CAP2),# data = temp_adapt_bioplot_data_asv, linetype = "solid",# arrow = arrow(length = unit(0.3, "cm")), size = 0.2,# color = "#676767") +#geom_text(data = temp_adapt_bioplot_data_asv, # aes(x = CAP1, y = CAP2, label = parameters), size = 2.5,# nudge_x = 0.05, nudge_y = 0.05) +theme_classic(base_size =12) +labs(title ="Capscale Analysis",subtitle ="Temperature Adaptation",x = cpa1_lab, y = cpa2_lab)temp_adapt_plot <- temp_adapt_plot +coord_fixed() +theme(aspect.ratio=1)temp_adapt_plotpng("files/metadata/figures/its18_temp_adapt_capscale.png",height =16, width =20, units ='cm', res =600, bg ="white")temp_adapt_plotinvisible(dev.off())pdf("files/metadata/figures/its18_temp_adapt_capscale.pdf",height =5, width =6)temp_adapt_plotdev.off()```</details>```{r}#| echo: falserm(list =ls(pattern ="tmp_"))```## Capscale Plots::: {.column-body}::: {.panel-tabset}#### Edaphic properties```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5edaphic_plot```<small>`r caption_fig_its("its_cap_edaph_plots")`</small>#### Soil Functional Response```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5soil_funct_plot```<small>`r caption_fig_its("its_cap_funct_plots")`</small>#### Temperature Adaptation```{r}#| echo: false#| eval: true#| warning: false#| fig-height: 4.5temp_adapt_plot```<small>`r caption_fig_its("its_cap_temp_plots")`</small>::::::```{r}#| echo: falseits18_capscale_plots <-ggarrange( edaphic_plot, soil_funct_plot, temp_adapt_plot,ncol =3, nrow =1, common.legend =TRUE, legend ="bottom")its18_capscale_plotsdev.off()png("files/metadata/figures/its18_capscale_plots.png", height =12, width =36,units ='cm', res =600, bg ="white")its18_capscale_plotsdev.off()pdf("files/metadata/figures/its18_capscale_plots.pdf", height =6, width =18)its18_capscale_plotsdev.off()``````{r}#| echo: false################################################### CODE FOR SUMMARY DATA ######################################################### Edaphicbioenv_set <- edaphic_bioenv_ind_mantelenvfit_set <- envfit_edaphic_mdres_name <-"its18_edaphic_sig_summary"metadata_set <-"edaphic"## BIOENVbioenv_tab <-NULLfor (i innames(bioenv_set)) { tmp_r <-data.frame(bioenv_set[[i]]$statistic)row.names(tmp_r) <- i tmp_p <-data.frame(bioenv_set[[i]]$signif)row.names(tmp_p) <- i bioenv_tab <-rbind(bioenv_tab, data.frame(tmp_r, tmp_p))rm(list =ls(pattern ="tmp_"))}bioenv_tab <- bioenv_tab %>% dplyr::rename("r (bioenv)"=1, "pval (bioenv)"=2) %>% tibble::rownames_to_column("parameter")## ENVFITtmp_r <-data.frame(envfit_set$vectors$r)tmp_r <- tmp_r %>% tibble::rownames_to_column("parameter")tmp_p <-data.frame(envfit_set$vectors$pvals)tmp_p <- tmp_p %>% tibble::rownames_to_column("parameter")tmp_rp <- dplyr::full_join(tmp_r, tmp_p, by ="parameter")tmp_rp <- tmp_rp %>% dplyr::rename("r2 (envfit)"=2, "Pr(>r) (envfit)"=3)tmp_rp <- base::subset(tmp_rp, c(tmp_rp$`Pr(>r)`<0.05& tmp_rp$r2 >0.4))tmp_summary <- dplyr::full_join(tmp_rp, bioenv_tab, by ="parameter") tmp_summary$metadata_set <- metadata_settmp_summary <- tmp_summary[, c(6,1:5)]assign(res_name, tmp_summary)rm(list =ls(pattern ="tmp_"))``````{r}#| echo: false## SOIL_FUNCTbioenv_set <- soil_funct_bioenv_ind_mantelenvfit_set <- envfit_soil_funct_mdres_name <-"its18_soil_funct_sig_summary"metadata_set <-"functional_response"## BIOENVbioenv_tab <-NULLfor (i innames(bioenv_set)) { tmp_r <-data.frame(bioenv_set[[i]]$statistic)row.names(tmp_r) <- i tmp_p <-data.frame(bioenv_set[[i]]$signif)row.names(tmp_p) <- i bioenv_tab <-rbind(bioenv_tab, data.frame(tmp_r, tmp_p))rm(list =ls(pattern ="tmp_"))}bioenv_tab <- bioenv_tab %>% dplyr::rename("r (bioenv)"=1, "pval (bioenv)"=2) %>% tibble::rownames_to_column("parameter")## ENVFITtmp_r <-data.frame(envfit_set$vectors$r)tmp_r <- tmp_r %>% tibble::rownames_to_column("parameter")tmp_p <-data.frame(envfit_set$vectors$pvals)tmp_p <- tmp_p %>% tibble::rownames_to_column("parameter")tmp_rp <- dplyr::full_join(tmp_r, tmp_p, by ="parameter")tmp_rp <- tmp_rp %>% dplyr::rename("r2 (envfit)"=2, "Pr(>r) (envfit)"=3)tmp_rp <- base::subset(tmp_rp, c(tmp_rp$`Pr(>r)`<0.05& tmp_rp$r2 >0.4))tmp_summary <- dplyr::full_join(tmp_rp, bioenv_tab, by ="parameter") tmp_summary$metadata_set <- metadata_settmp_summary <- tmp_summary[, c(6,1:5)]assign(res_name, tmp_summary)rm(list =ls(pattern ="tmp_"))``````{r}#| echo: false## TEMPbioenv_set <- temp_adapt_bioenv_ind_mantelenvfit_set <- envfit_temp_adapt_mdres_name <-"its18_temp_adapt_sig_summary"metadata_set <-"temperature adaptation"## BIOENVbioenv_tab <-NULLfor (i innames(bioenv_set)) { tmp_r <-data.frame(bioenv_set[[i]]$statistic)row.names(tmp_r) <- i tmp_p <-data.frame(bioenv_set[[i]]$signif)row.names(tmp_p) <- i bioenv_tab <-rbind(bioenv_tab, data.frame(tmp_r, tmp_p))rm(list =ls(pattern ="tmp_"))}bioenv_tab <- bioenv_tab %>% dplyr::rename("r (bioenv)"=1, "pval (bioenv)"=2) %>% tibble::rownames_to_column("parameter")## ENVFITtmp_r <-data.frame(envfit_set$vectors$r)tmp_r <- tmp_r %>% tibble::rownames_to_column("parameter")tmp_p <-data.frame(envfit_set$vectors$pvals)tmp_p <- tmp_p %>% tibble::rownames_to_column("parameter")tmp_rp <- dplyr::full_join(tmp_r, tmp_p, by ="parameter")tmp_rp <- tmp_rp %>% dplyr::rename("r2 (envfit)"=2, "Pr(>r) (envfit)"=3)tmp_rp <- base::subset(tmp_rp, c(tmp_rp$`Pr(>r)`<0.05& tmp_rp$r2 >0.4))tmp_summary <- dplyr::full_join(tmp_rp, bioenv_tab, by ="parameter") tmp_summary$metadata_set <- metadata_settmp_summary <- tmp_summary[, c(6,1:5)]assign(res_name, tmp_summary)rm(list =ls(pattern ="tmp_"))rm(bioenv_set, envfit_set, res_name, metadata_set, bioenv_tab)its18_sig_summary <-rbind(its18_edaphic_sig_summary, its18_soil_funct_sig_summary, its18_temp_adapt_sig_summary)rm(its18_edaphic_sig_summary, its18_soil_funct_sig_summary, its18_temp_adapt_sig_summary)``````{r}#| echo: falsesave.image("page_build/metadata_its18_wf.rdata")gdata::keep(ssu18_sig_summary, its18_sig_summary, sure =TRUE)save.image("page_build/metadata_summary.rdata")``````{r}#| include: false#| eval: trueremove(list =ls())```# Workflow Output Data products generated in this workflow can be downloaded from figshare.<iframe src="https://widgets.figshare.com/articles/16828294/embed?show_title=1" width="100%" height="251" allowfullscreen frameborder="0"></iframe></br><a href="da.html" class="btnnav button_round" style="float: left;">Previous workflow:<br> 7. Differentially Abundant ASVs & Taxa</a><br><br>```{r}#| message: false #| results: hide#| eval: true#| echo: falseremove(list =ls())source(file.path("assets", "functions.R"))```#### Source Code {.appendix}The source code for this page can be accessed on GitHub `r fa(name = "github")` by [clicking this link](`r source_code()`). #### Data Availability {.appendix}Data generated in this workflow and the Rdata need to run the workflow can be accessed on figshare at [10.25573/data.16828294](https://doi.org/10.25573/data.16828294).#### Last updated on {.appendix}```{r}#| echo: false#| eval: trueSys.time()```