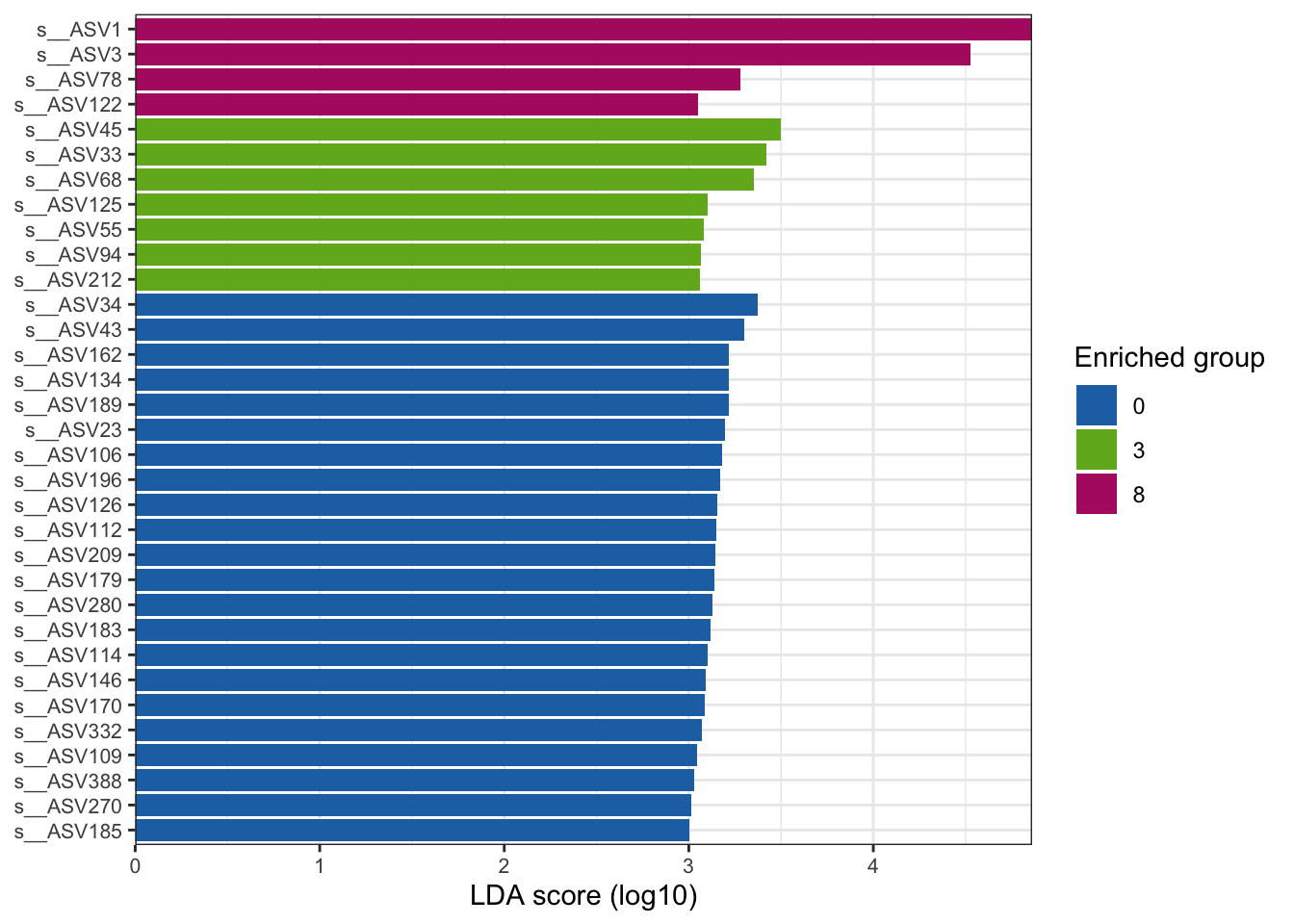
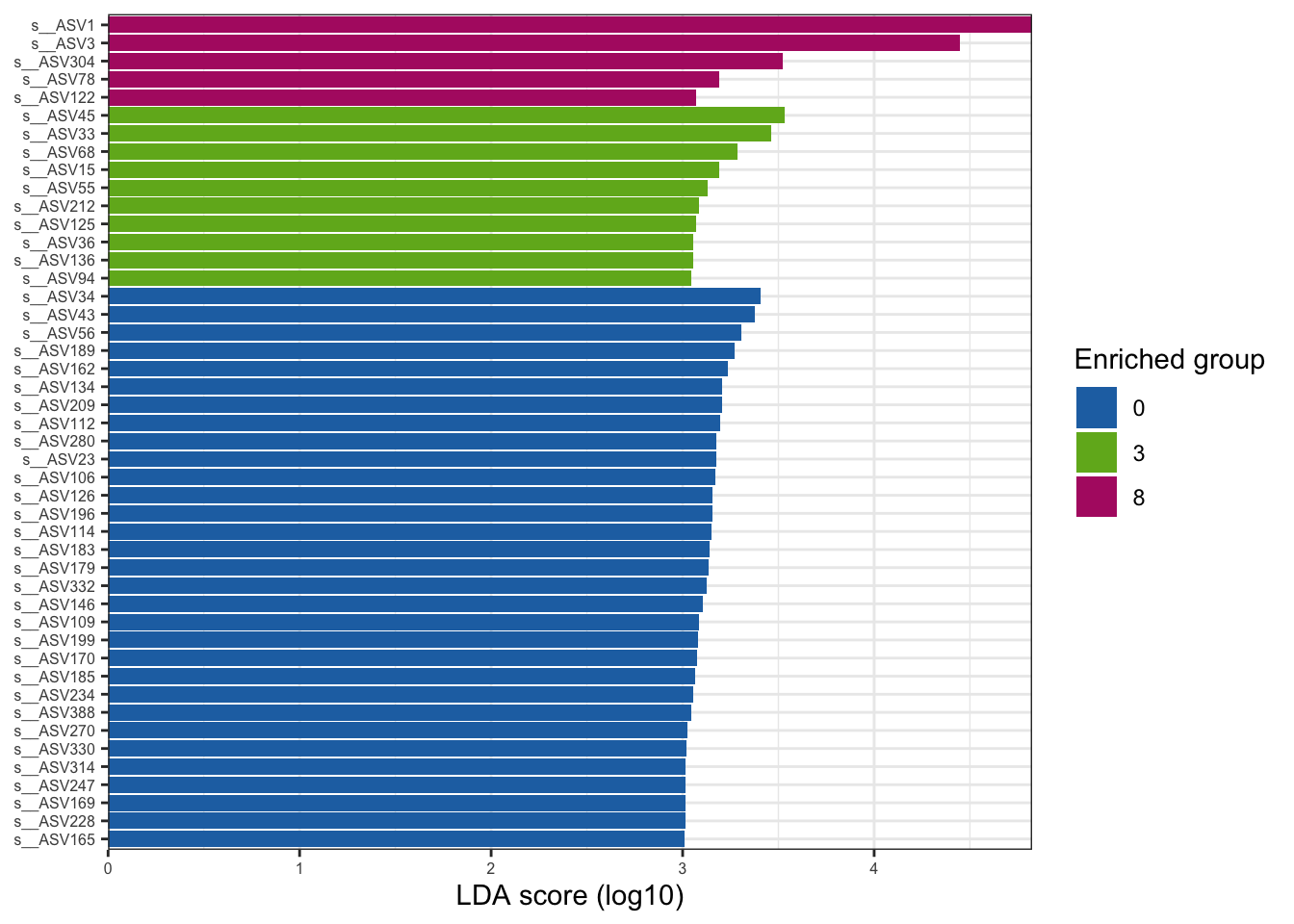
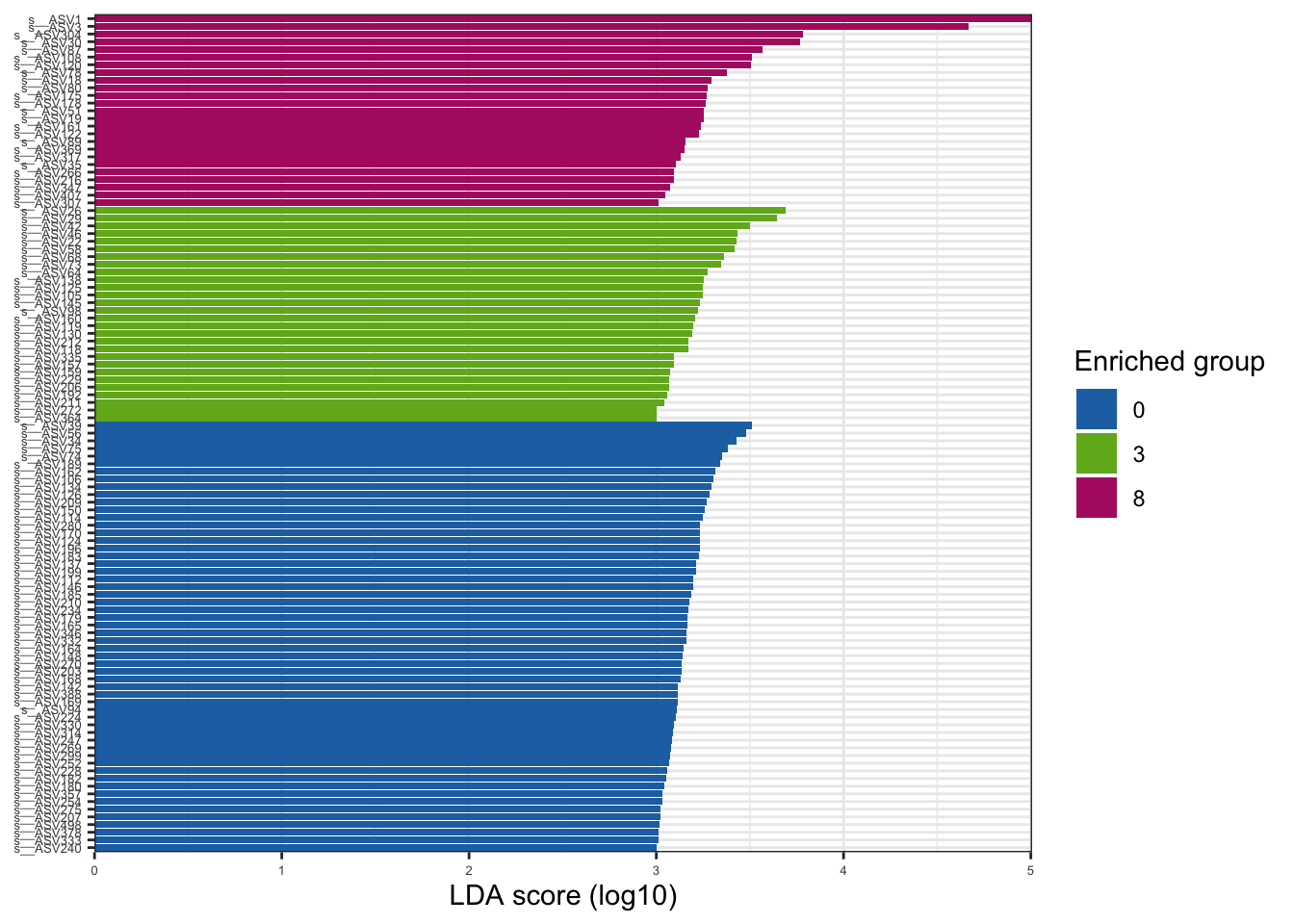
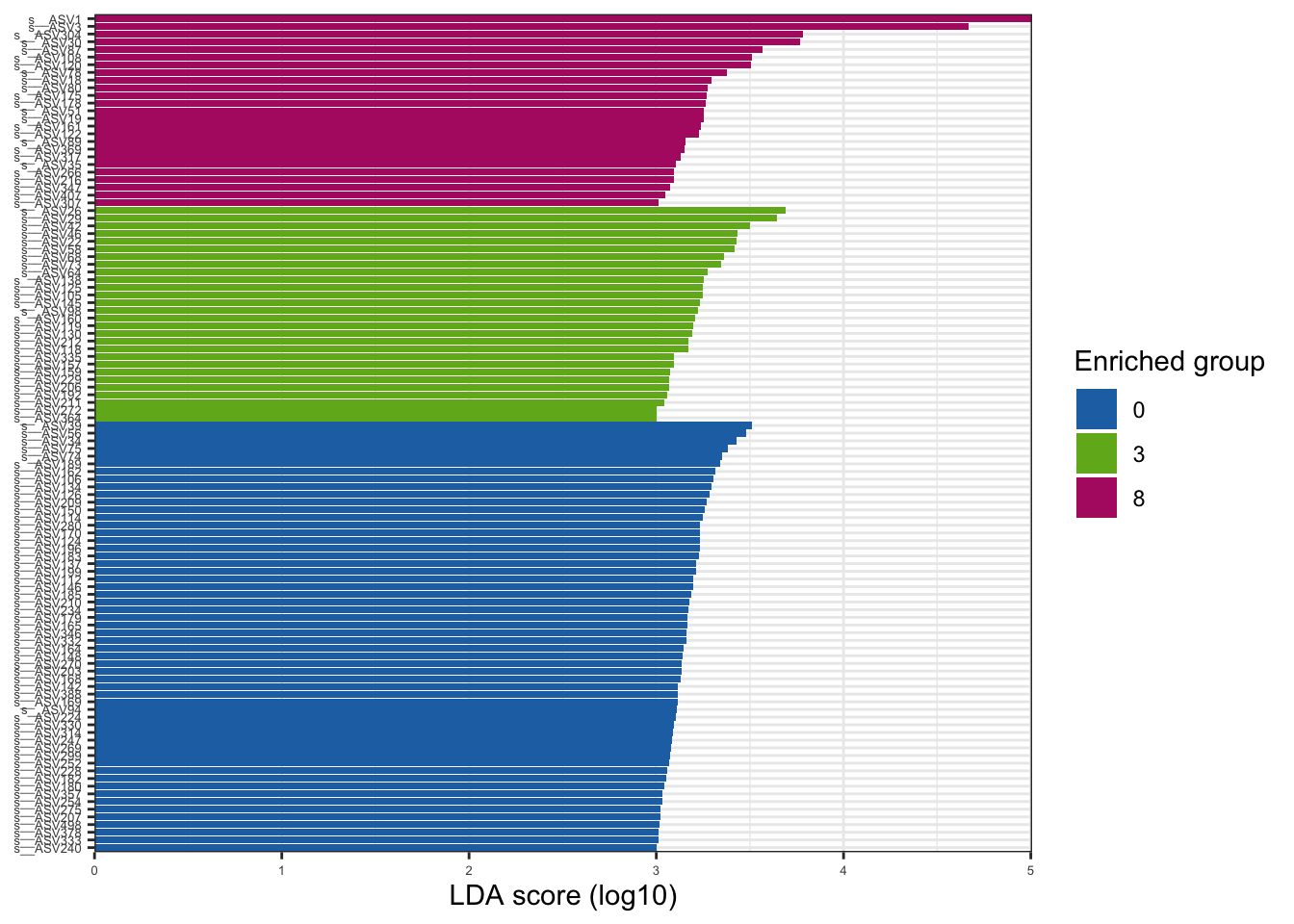
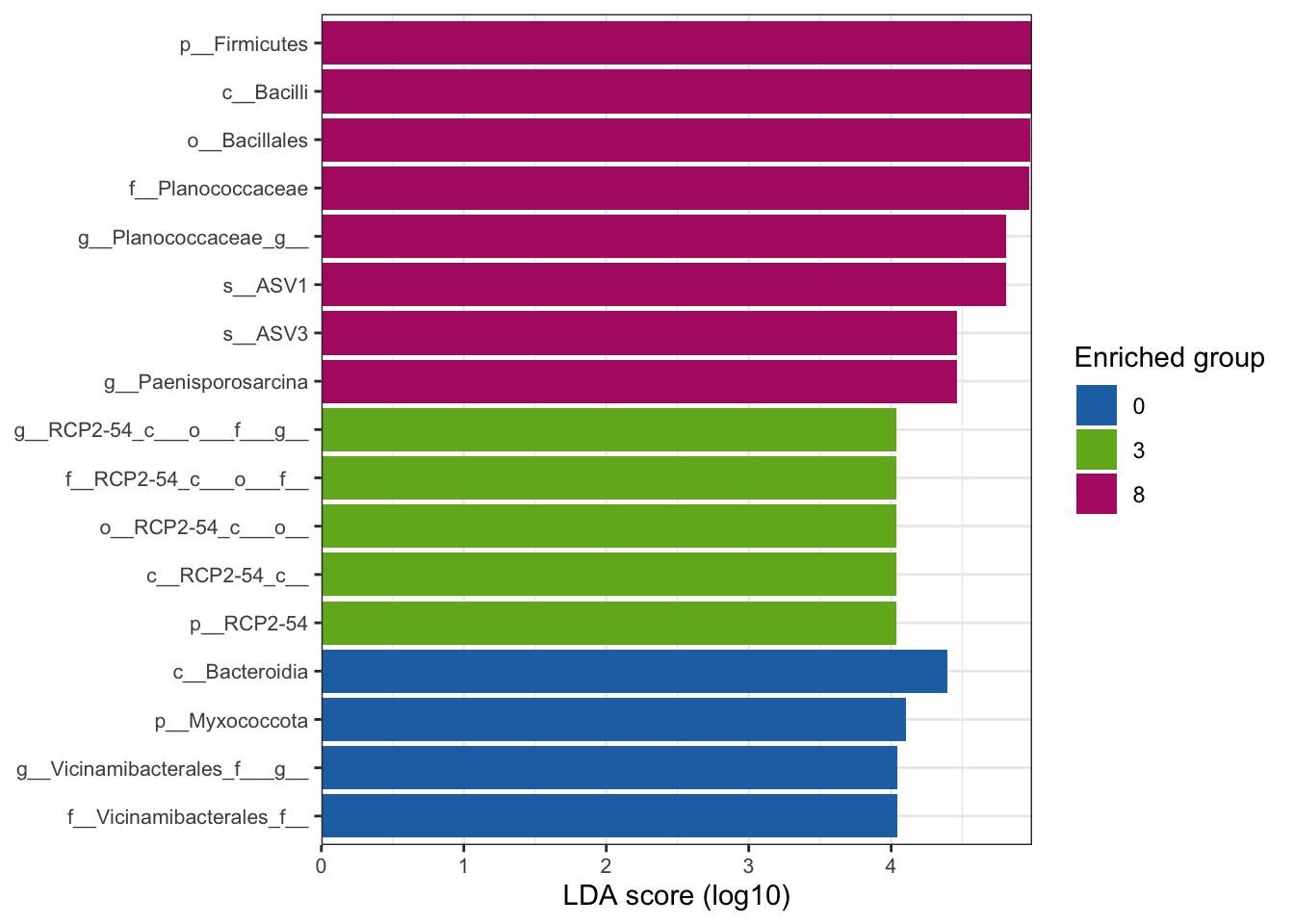
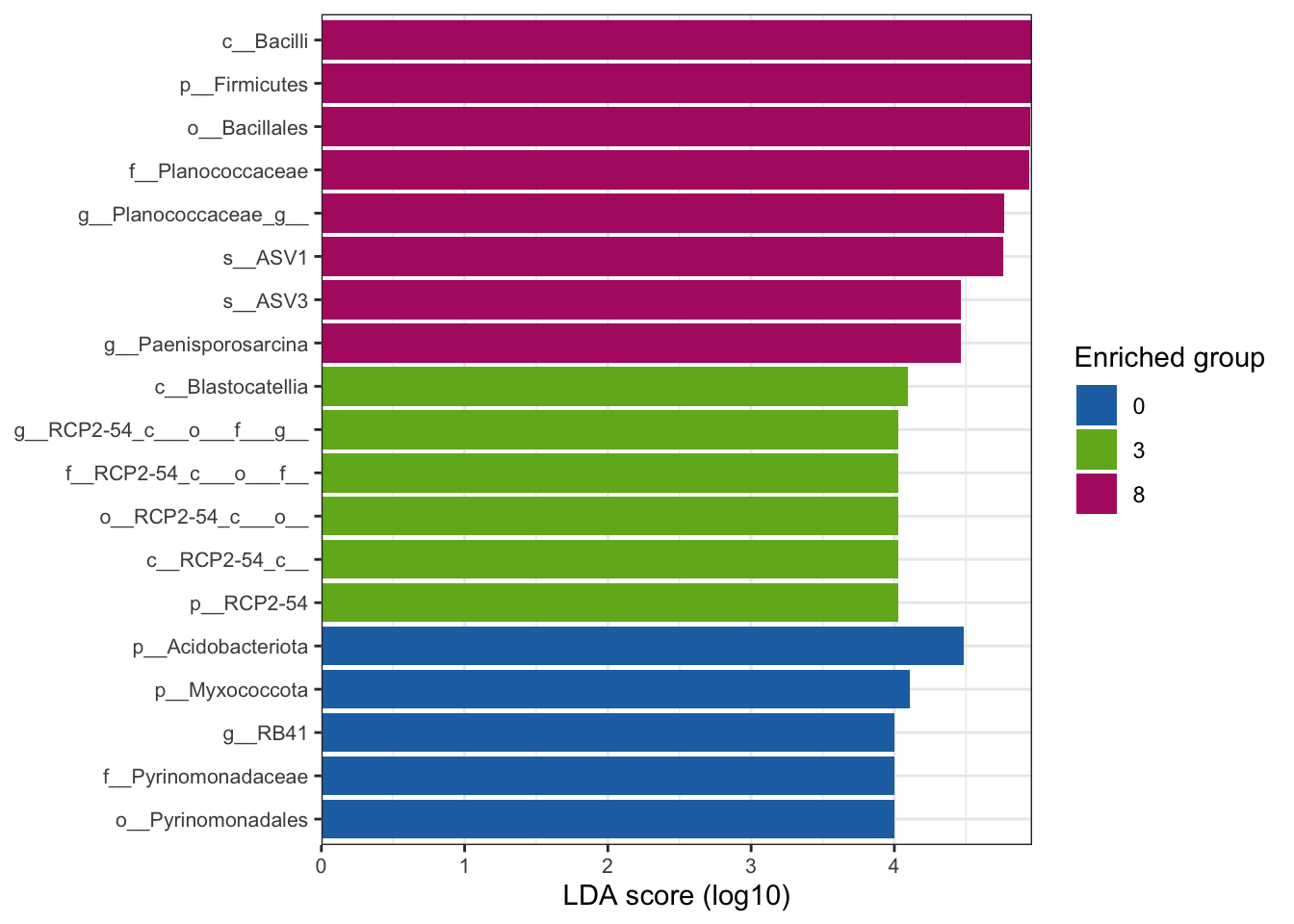
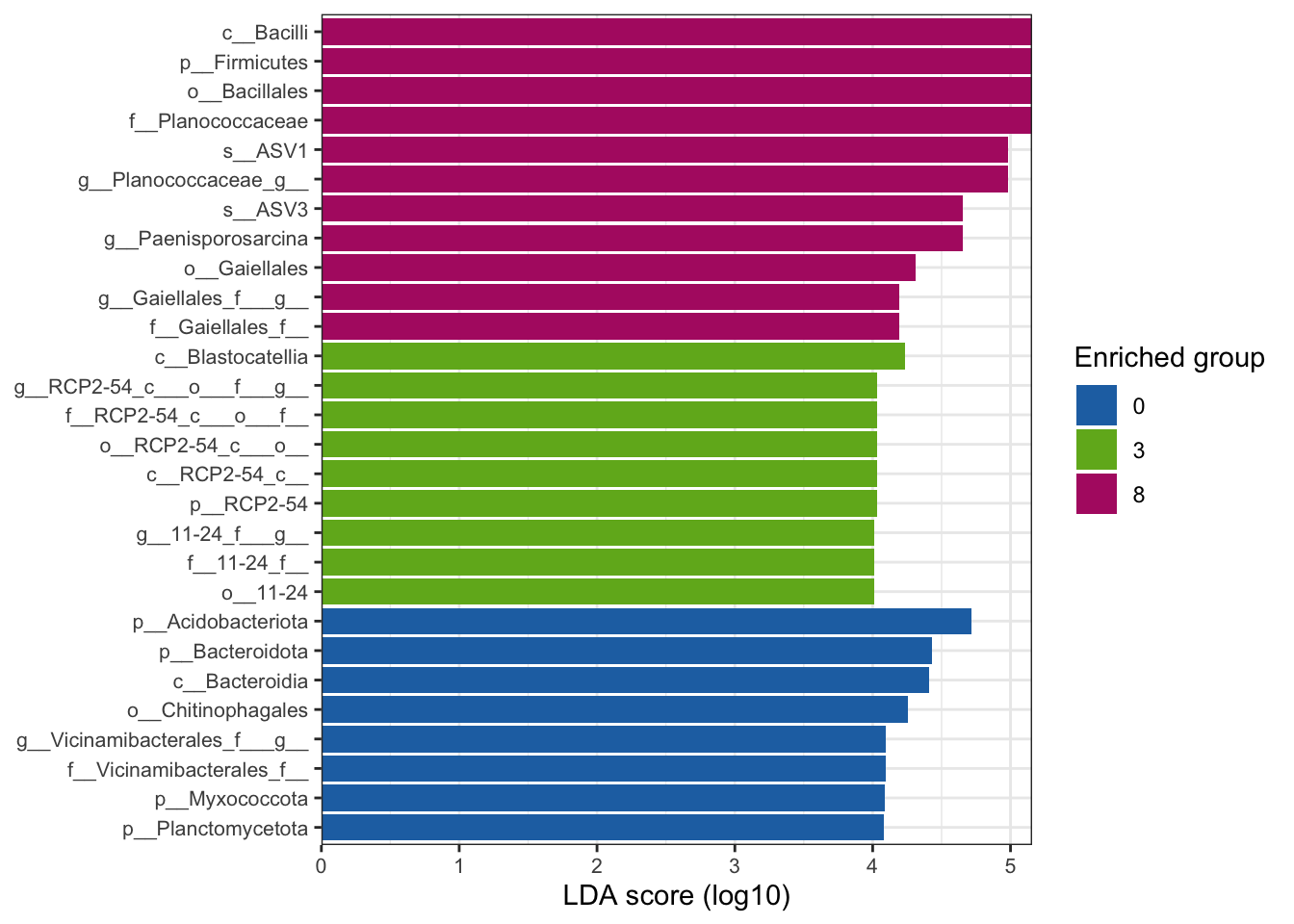
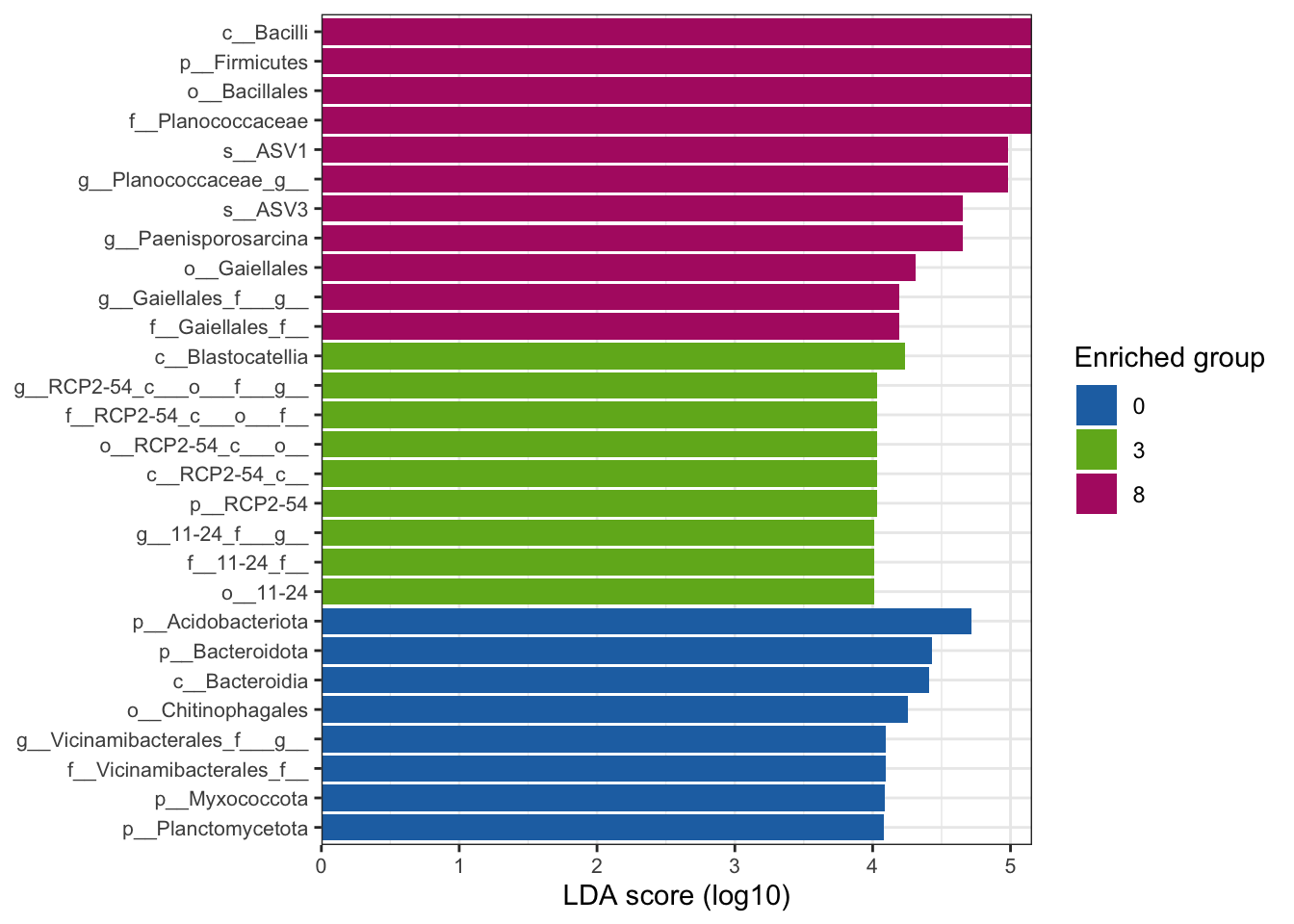
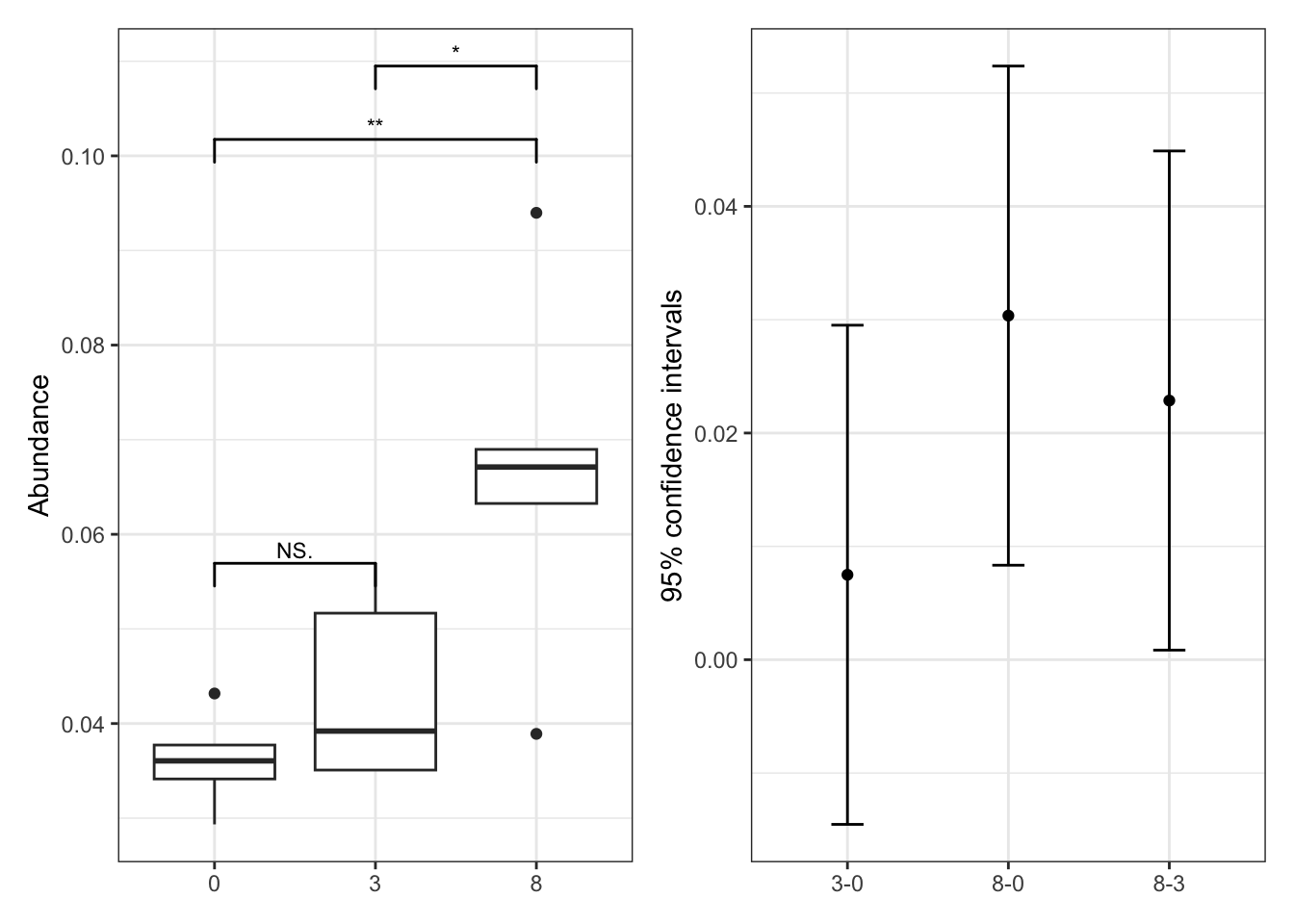
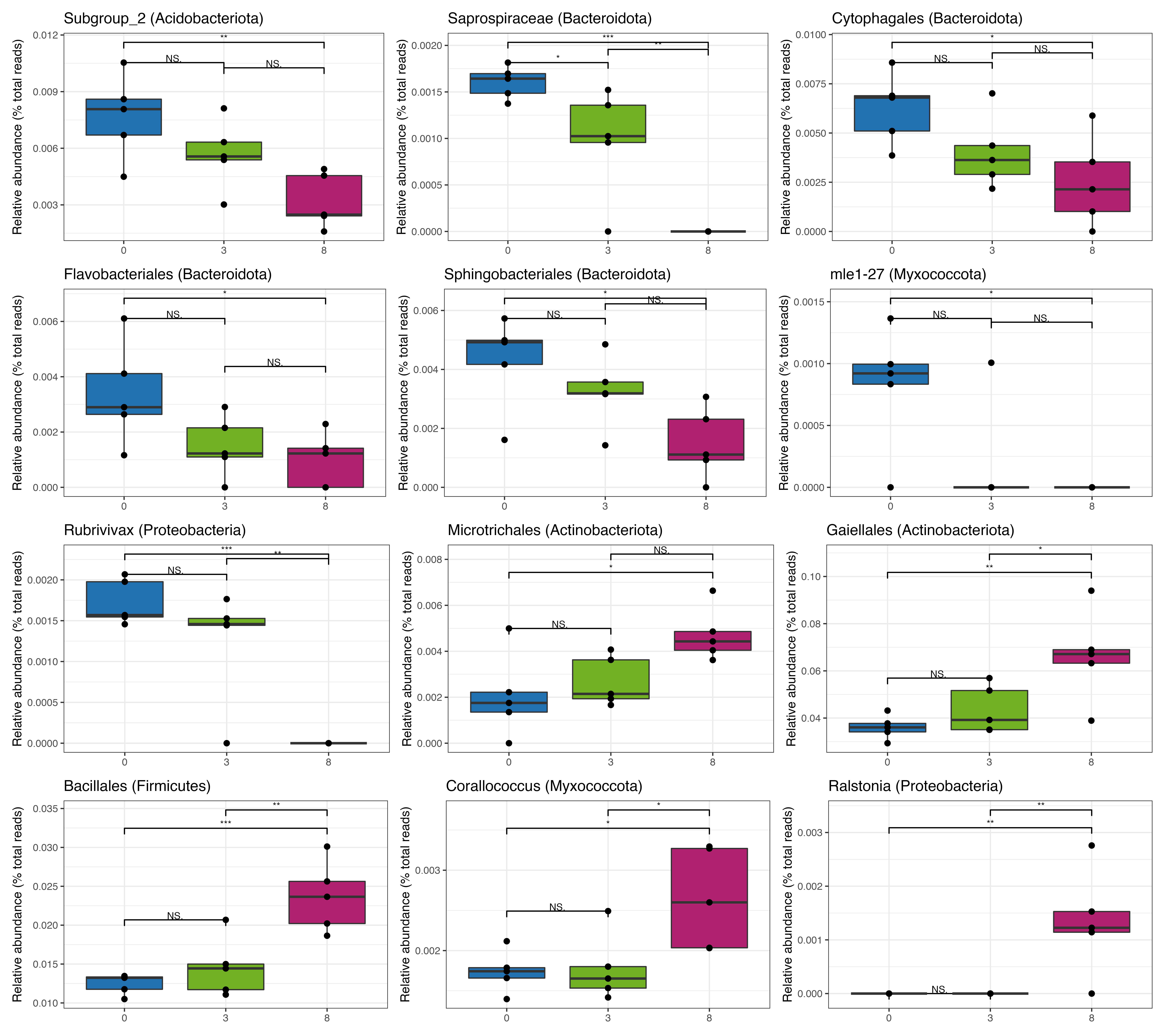
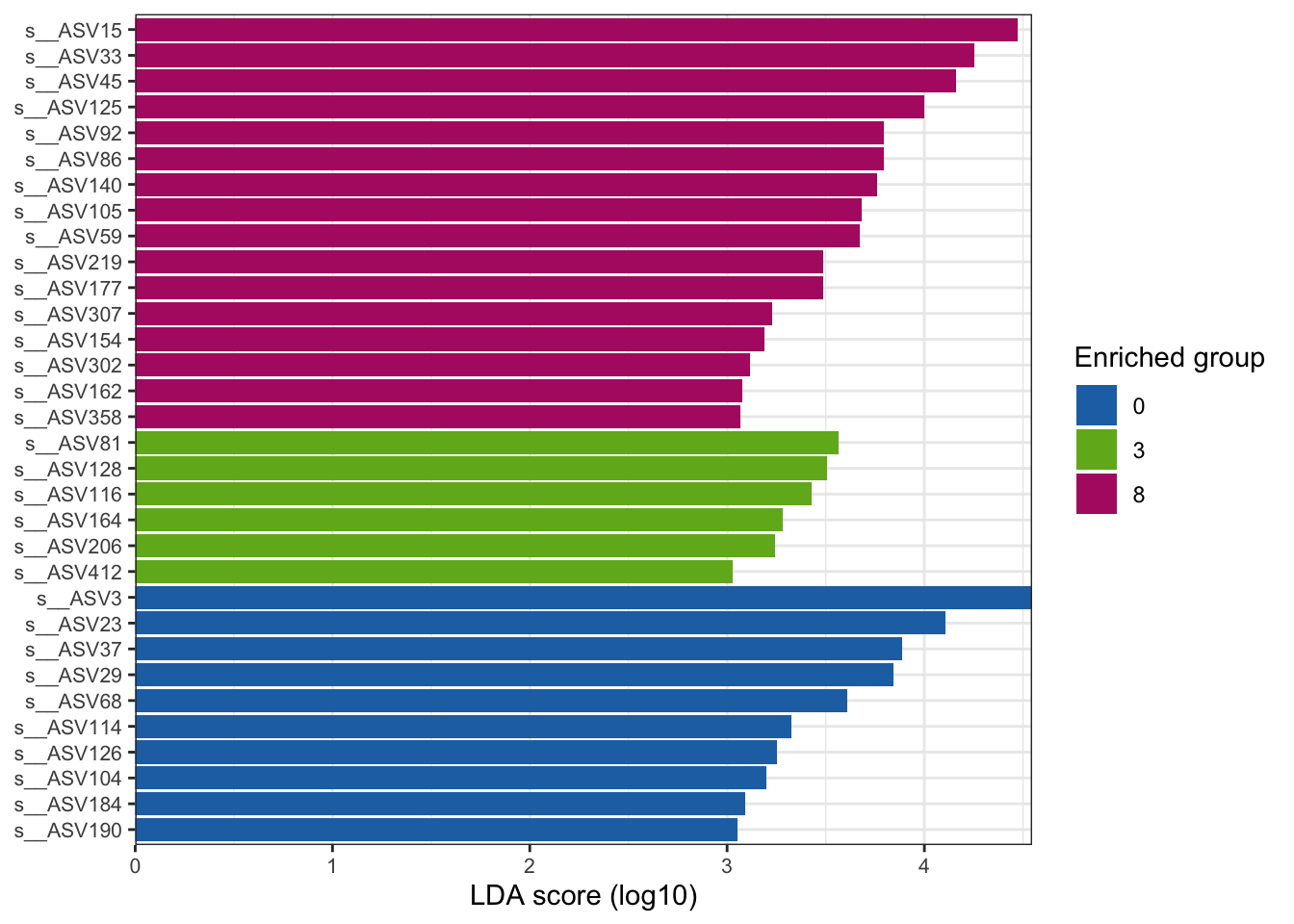
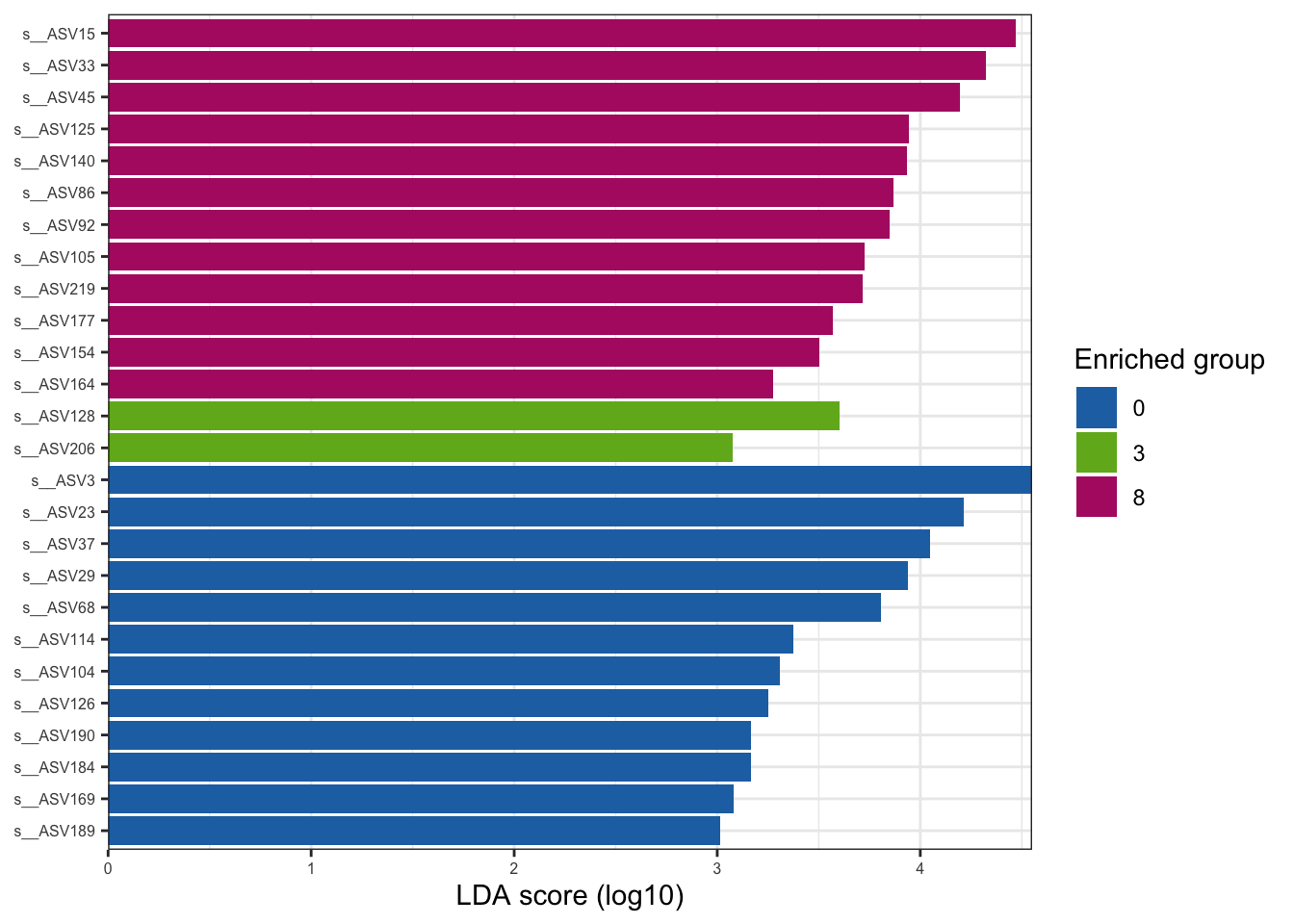
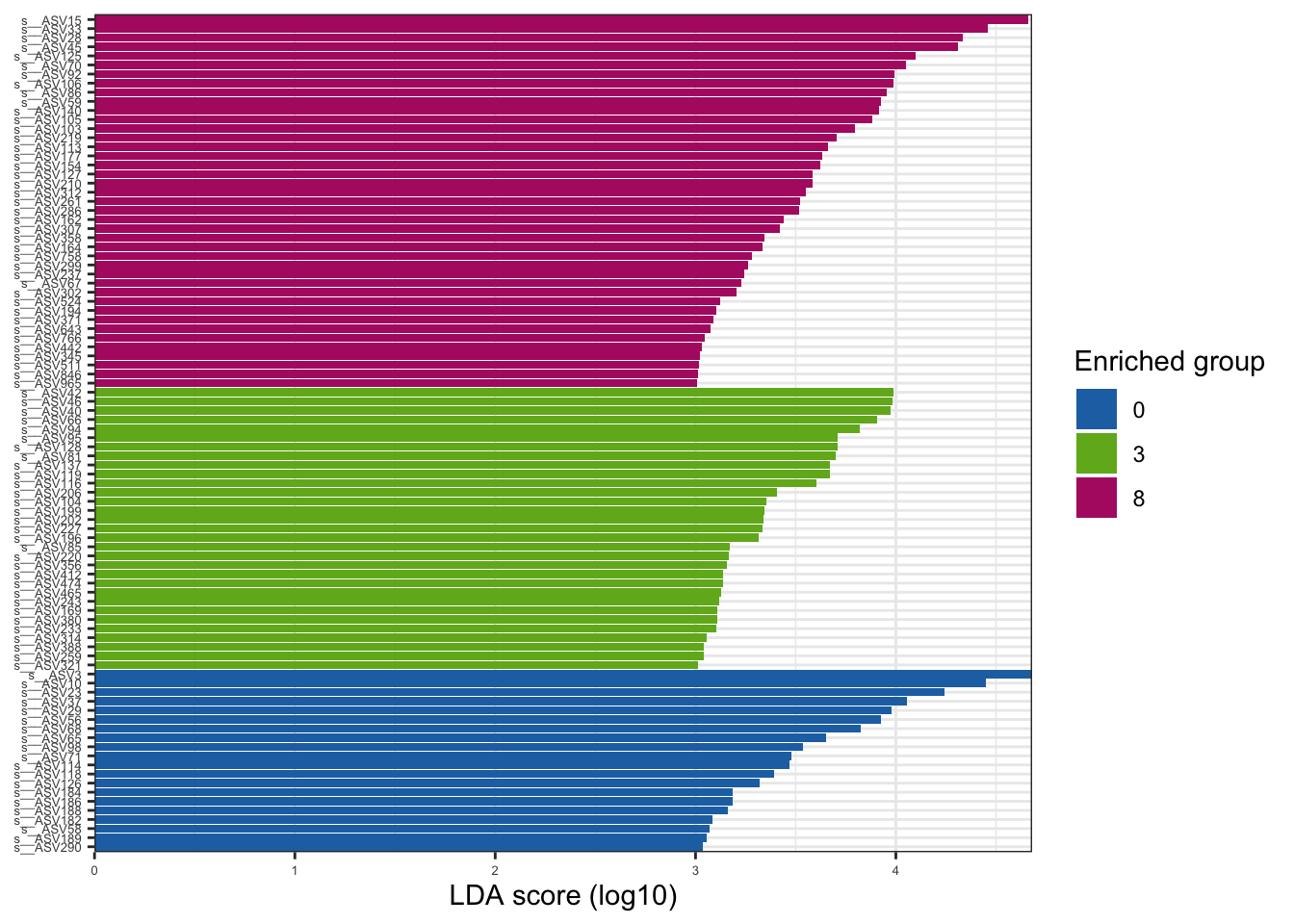
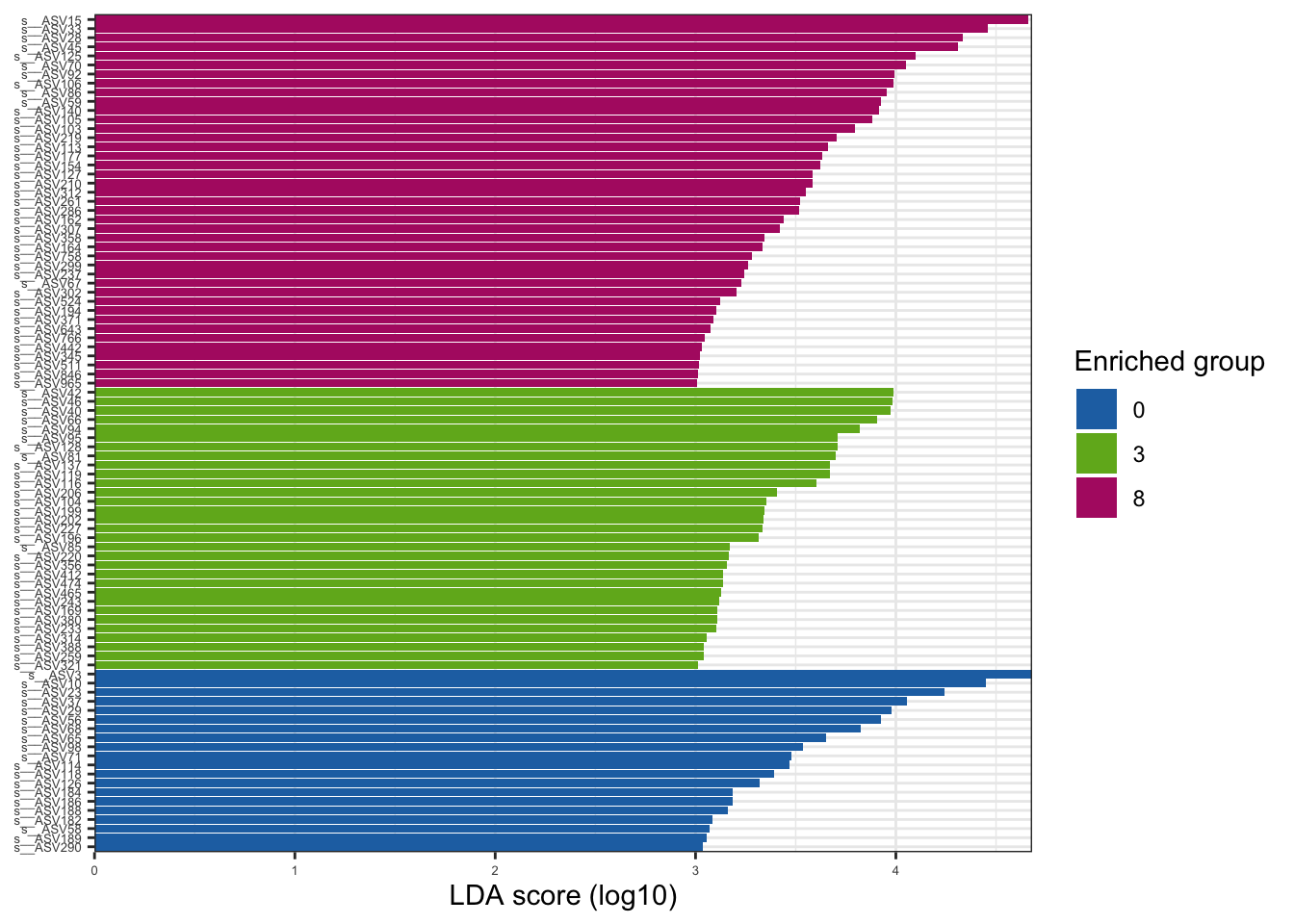
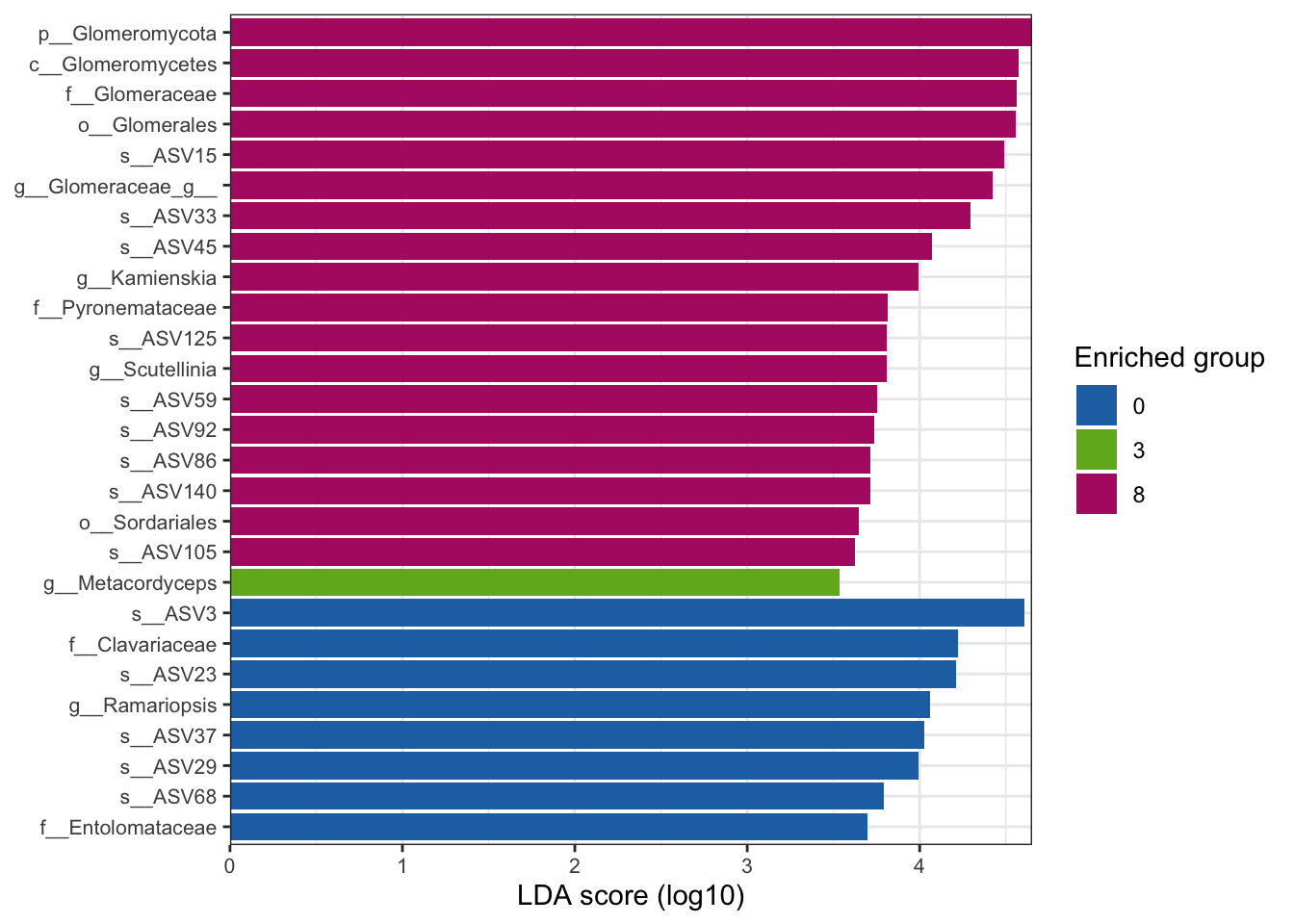
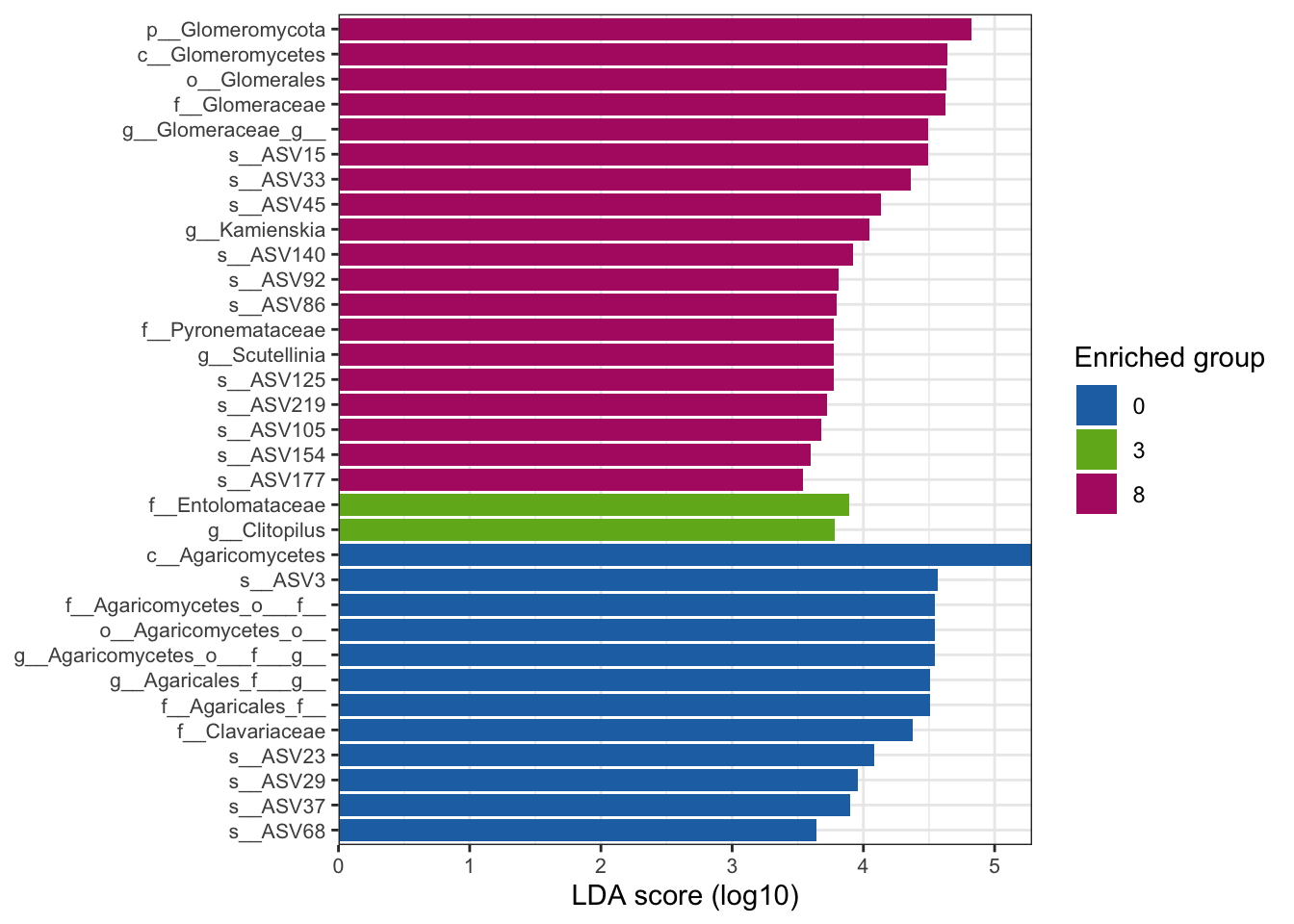
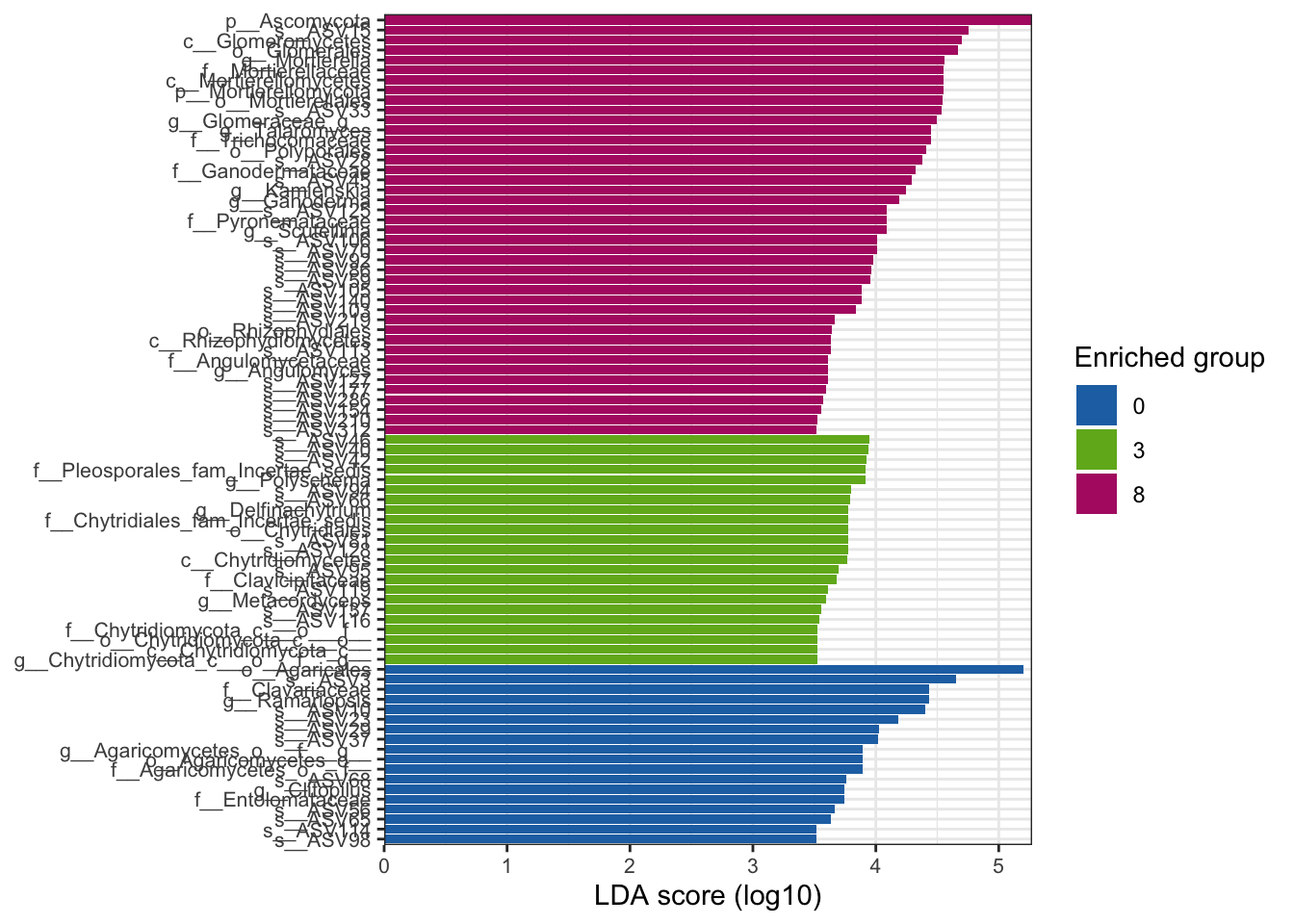
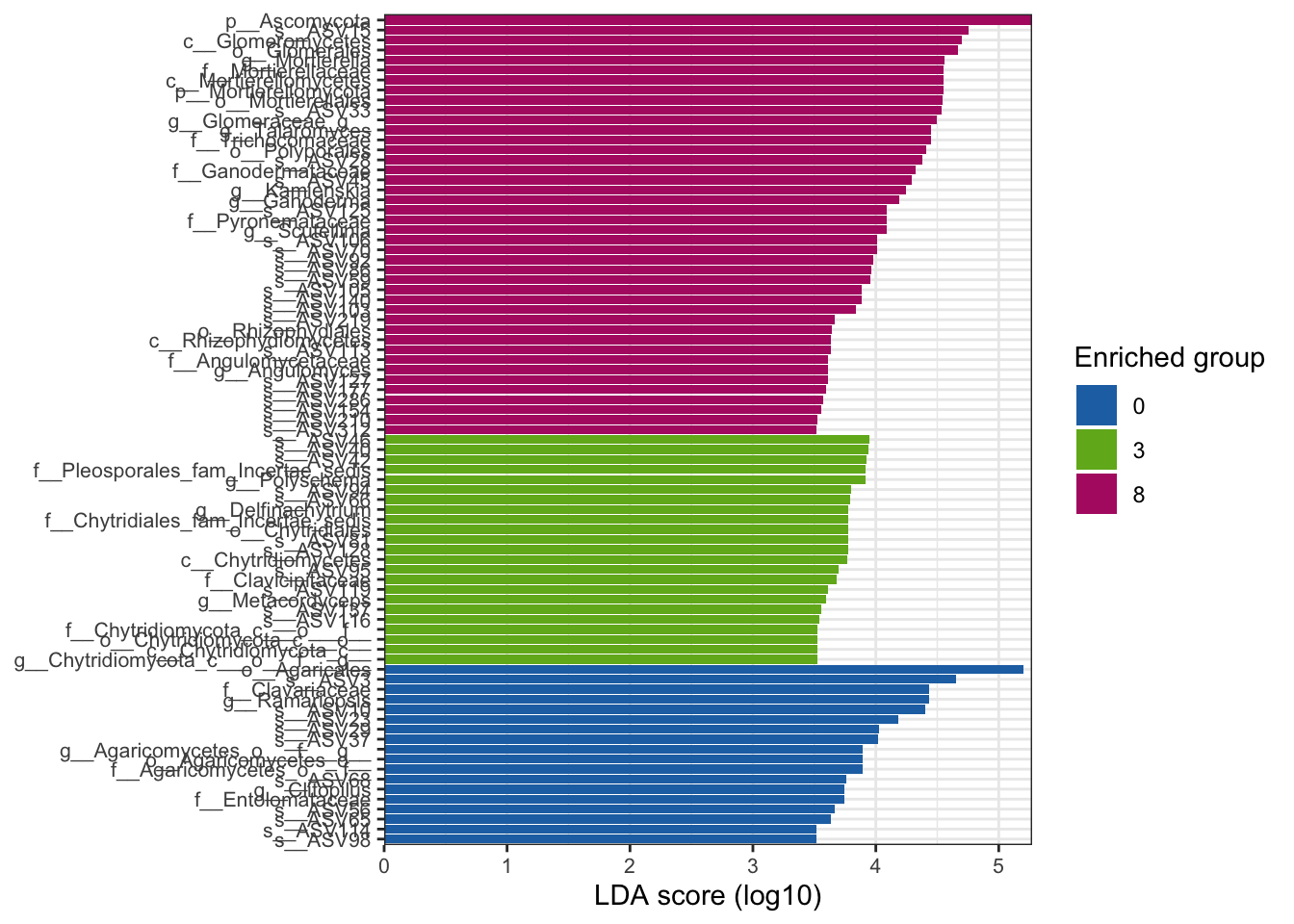
--- title: "7. Differentially Abundant ASVs & Taxa" description: | Reproducible workflow to assess differentially abundant ASVs and taxa between temperature treatments using Indicator Species Analysis and linear discriminant analysis (LDA) effect size (LEfSe). format: html: toc: true toc-depth: 2 --- ```{r} #| message: false #| results: hide #| code-fold: true #| code-summary: "Click here for setup information" :: opts_chunk$ set (echo = TRUE , eval = FALSE )set.seed (119 )#library(conflicted) #pacman::p_depends(microbiomeMarker, local = TRUE) #pacman::p_depends_reverse(microbiomeMarker, local = TRUE) library (phyloseq); packageVersion ("phyloseq" )library (Biostrings); packageVersion ("Biostrings" ):: p_load (tidyverse, patchwork, ampvis2, install = FALSE , update = FALSE )options (scipen= 999 ):: opts_current$ get (c ("cache" ,"cache.path" ,"cache.rebuild" ,"dependson" ,"autodep" ``` ```{r} #| include: false #| eval: true ## Load to build page only #2 remove (list = ls ())load ("page_build/da_wf.rdata" )load ("page_build/mmarker_wf.rdata" )objects ()``` ```{r} #| echo: false #| eval: true #| results: hide # Create the caption(s) with captioner <- captioner (prefix = "(16S rRNA) Table" , suffix = " |" , style = "b" )<- captioner (prefix = "(16S rRNA) Figure" , suffix = " |" , style = "b" )<- captioner (prefix = "(ITS) Table" , suffix = " |" , style = "b" )<- captioner (prefix = "(ITS) Figure" , suffix = " |" , style = "b" )# Create a function for referring to the tables in text <- function (x) str_extract (x, "[^|]*" ) %>% trimws (which = "right" , whitespace = "[ ]" )``` ```{r} #| echo: false #| eval: true source ("assets/captions/captions_da.R" )``` # Synopsis [ Data Preparation workflow ](data-prep.html) and the [ Filtering workflow ](filtering.html) or or download the files linked below. See the [ Data Availability ](data-availability.html) page for complete details.## Workflow Input < iframe src = "https://widgets.figshare.com/articles/14690739/embed?show_title=1" width = "100%" height = "251" allowfullscreen frameborder = "0" ></ iframe > < iframe src = "https://widgets.figshare.com/articles/14701440/embed?show_title=1" width = "100%" height = "251" allowfullscreen frameborder = "0" ></ iframe > `labdsv` package [ @roberts2016package ] to run Dufrene-Legendre Indicator Species Analysis and the [ microbiomeMarker package ](https://github.com/yiluheihei/microbiomeMarker) [ @cao2020package ] to run linear discriminant analysis (LDA) effect size (LEfSe) [ @segata2011metagenomic ] .# 16s rRNA ```{r} #| include: false #| eval: false ## Initial Load for ANALYSIS #1 remove (list = ls ())<- readRDS ("files/data-prep/rdata/ssu18_ps_work.rds" )<- readRDS ("files/filtering/arbitrary/rdata/ssu18_ps_filt.rds" )<- readRDS ("files/filtering/perfect/rdata/ssu18_ps_perfect.rds" )<- readRDS ("files/filtering/pime/rdata/ssu18_ps_pime.rds" )``` ```{r} #| echo: false <- c ("ssu18_ps_work" , "ssu18_ps_filt" , "ssu18_ps_perfect" , "ssu18_ps_pime" ) for (i in samp_ps) {<- get (i)<- file.path ("files/alpha/rdata/" )<- readRDS (paste (tmp_path, i, ".rds" , sep = "" ))<- sample_data (tmp_read)<- data.frame (tax_table (tmp_get))#tmp_tax_data[,c(1:6)] $ ASV_ID <- NULL # Some have, some do not <- data.frame (lapply (tmp_tax_data, gsub, pattern = "^[k | p | c | o | f]_.*" , replacement = NA , fixed = FALSE ))<- as.matrix (tmp_tax_data)<- phyloseq (otu_table (tmp_get),phy_tree (tmp_get),tax_table (tmp_tax_data),assign (i, tmp_ps)rm (list = ls (pattern = "tmp_" ))objects ()``` ```{r} #| echo: false <- c ("#2271B2" , "#71B222" , "#B22271" )``` ## Indicator Analysis 1. Choose a data set(s) & set the p-value cutoff.```{r} #| code-fold: true <- c ("ssu18_ps_work" , "ssu18_ps_filt" , "ssu18_ps_perfect" , "ssu18_ps_pime" )<- 0.05 ``` 2. Format Data Frame```{r} #| code-fold: true for (i in samp_ps) {<- get (i)<- data.frame (otu_table (tmp_get))<- tmp_df[, which (colSums (tmp_df) != 0 )]<- row.names (tmp_df)<- tmp_row_names %>% :: str_replace ("P[0-9]{2}_D[0-9]{2}_[0-9]{3}_" , "" ) %>% :: str_replace ("[A-Z]$" , "" )<- tmp_df %>% tibble:: add_column (tmp_row_names, .before = 1 )<- purrr:: map_chr (i, ~ paste0 (., "_seq_tab" ))assign (tmp_name, tmp_df)rm (list = ls (pattern = "tmp_" ))objects ()``` 3. Run Indicator Analysis```{r} #| code-fold: true set.seed (1191 )for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_seq_tab" )))<- indval (tmp_get[,- 1 ], tmp_get[,1 ])<- tmp_iva$ maxcls[tmp_iva$ pval <= indval_pval]<- tmp_iva$ indcls[tmp_iva$ pval <= indval_pval]<- tmp_iva$ pval[tmp_iva$ pval <= indval_pval]<- apply (tmp_get[,- 1 ] > 0 , 2 , sum)[tmp_iva$ pval <= indval_pval]<- data.frame (group = tmp_gr, indval = tmp_iv,pval = tmp_pv, freq = tmp_fr)<- tmp_sum[order (tmp_sum$ group, - tmp_sum$ indval),]<- data.frame (tax_table (get (i)))$ ASV_ID <- NULL <- merge (tmp_sum, tmp_tax_df, by = "row.names" , all = TRUE )<- tmp_sum_tax[! (is.na (tmp_sum_tax$ group)),]class (tmp_sum_tax$ group) <- "character" $ group <- stringr:: str_replace (tmp_sum_tax$ group, "^1$" , "C0" )$ group <- stringr:: str_replace (tmp_sum_tax$ group, "^2$" , "W3" )$ group <- stringr:: str_replace (tmp_sum_tax$ group, "^3$" , "W8" )<- tmp_sum_tax %>% dplyr:: rename ("ASV_ID" = "Row.names" )<- tmp_sum_tax[order (as.numeric (gsub ("[A-Z]{3}" , "" , $ ASV_ID))),]$ ASV_ID <- as.character (tmp_sum_tax$ ASV_ID)<- p.adjust (tmp_sum$ pval, "bonferroni" )<- purrr:: map_chr (i, ~ paste0 (., "_indval_summary" ))assign (tmp_res_name, tmp_sum_tax)rm (list = ls (pattern = "tmp_" ))objects ()``` ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_seq_tab" )))1 ] <- NULL <- as.data.frame (t (tmp_get))<- colnames (tmp_df)<- tmp_col_names %>% :: str_replace ("P[0-9]{2}_D[0-9]{2}_[0-9]{3}_" , "" ) %>% :: str_replace ("[A-Z]$" , "" )colnames (tmp_df) <- tmp_col_names#tmp_df$freq_all <- apply(tmp_df > 0, 1, sum) $ freq_C0 <- apply (tmp_df[ , (names (tmp_df) %in% "C0" )] > 0 , 1 , sum)$ freq_W3 <- apply (tmp_df[ , (names (tmp_df) %in% "W3" )] > 0 , 1 , sum)$ freq_W8 <- apply (tmp_df[ , (names (tmp_df) %in% "W8" )] > 0 , 1 , sum)$ reads_total <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% c ("C0" , "W3" , "W8" ))])$ reads_C0 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "C0" )])$ reads_W3 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "W3" )])$ reads_W8 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "W8" )])<- tmp_df[,! grepl ("^[C | W]" , names (tmp_df))]<- tmp_df %>% tibble:: rownames_to_column ("ASV_ID" )<- get (purrr:: map_chr (i, ~ paste0 (., "_indval_summary" )))<- merge (tmp_df, tmp_get_indval, by = "ASV_ID" , all = FALSE )<- tmp_merge_df[,c (1 ,9 : 12 ,2 : 8 ,13 : 19 )]<- tmp_merge_df[order (tmp_merge_df$ reads_total, decreasing = TRUE ), ]<- purrr:: map_chr (i, ~ paste0 (., "_indval_final" ))assign (tmp_merge_name, tmp_merge_df)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_indval_summary" )``` 4. Save a new phyloseq object```{r} #| code-fold: true for (i in samp_ps) {<- get (i)<- get (purrr:: map_chr (i, ~ paste0 (., "_indval_summary" )))<- tmp_tab[,1 ]<- prune_taxa (tmp_list, tmp_get)<- purrr:: map_chr (i, ~ paste0 (., "_ind" ))assign (tmp_name, tmp_ps)@ phy_tree <- NULL <- prune_samples (sample_sums (tmp_ps) > 0 , tmp_ps)<- rtree (ntaxa (tmp_ps), rooted = TRUE ,tip.label = taxa_names (tmp_ps))<- merge_phyloseq (tmp_ps, sample_data, tmp_ps_tree)assign (tmp_name, tmp_ps)rm (list = ls (pattern = "tmp_" ))objects ()``` ## Indicator Summary </ br > #### FULL ```{r} #| echo: false #| eval: true <- ssu18_ps_work_indval_finalwrite.table (seq_table, "files/da/tables/ssu_isa_results_full.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_isa_full")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),indval = colDef (name = "indval score" , align = "center" , filterable = FALSE ,minWidth = 130 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "Indicator analysis results" , sticky = "left" ,columns = c ("group" , "indval" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### Arbitrary filtered ```{r} #| echo: false #| eval: true <- ssu18_ps_filt_indval_finalwrite.table (seq_table, "files/da/tables/ssu_isa_results_arbitrary.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_isa_filt")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),indval = colDef (name = "indval score" , align = "center" , filterable = FALSE ,minWidth = 130 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "Indicator analysis results" , sticky = "left" ,columns = c ("group" , "indval" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PERfect filtered ```{r} #| echo: false #| eval: true <- ssu18_ps_perfect_indval_finalwrite.table (seq_table, "files/da/tables/ssu_isa_results_perfect.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_isa_perfect")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),indval = colDef (name = "indval score" , align = "center" , filterable = FALSE ,minWidth = 130 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "Indicator analysis results" , sticky = "left" ,columns = c ("group" , "indval" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PIME filtered ```{r} #| echo: false #| eval: true <- ssu18_ps_pime_indval_finalwrite.table (seq_table, "files/da/tables/ssu_isa_results_pime.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_isa_pime")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),indval = colDef (name = "indval score" , align = "center" , filterable = FALSE ,minWidth = 130 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "Indicator analysis results" , sticky = "left" ,columns = c ("group" , "indval" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` ## LEfSe Analysis [ @segata2011metagenomic ] .```{r} #| code-fold: true for (i in samp_ps) {<- get (i)tax_table (tmp_get) <- tax_table (tmp_get)[,c (1 : 6 )]tax_table (tmp_get) <- cbind (tax_table (tmp_get),rownames (tax_table (tmp_get)))colnames (tax_table (tmp_get)) <- c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" , "Species" )<- purrr:: map_chr (i, ~ paste0 (., "_rf_all" ))assign (tmp_name, tmp_get)tax_table (tmp_get) <- tax_table (tmp_get)[,c (7 )]<- purrr:: map_chr (i, ~ paste0 (., "_rf_asv" ))assign (tmp_name_asv, tmp_get)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_rf" )``` 1. Choose a data set(s) & set the LEfSe cutoff values.```{r} <- c ("ssu18_ps_work" , "ssu18_ps_filt" , "ssu18_ps_perfect" , "ssu18_ps_pime" )<- 2 <- 0.05 <- 0.05 ``` ```{r} #| warnings: false #| error: false #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_rf_asv" )))<- run_lefse (tmp_get, norm = "CPM" , class = "TEMP" , correct = "2" , lda_cutoff = lefse_lda, kw_cutoff = lefse_kw, wilcoxon_cutoff = lefse_wc, bootstrap_n = 30 , bootstrap_fraction = 2 / 3 , sample_min = 10 , multicls_strat = FALSE , curv = FALSE )<- purrr:: map_chr (i, ~ paste0 (., "_lefse_asv" ))assign (tmp_name, tmp_lefse)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_lefse_asv" )for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_rf_all" )))<- run_lefse (tmp_get, norm = "CPM" , class = "TEMP" , correct = "2" , lda_cutoff = lefse_lda, kw_cutoff = lefse_kw, wilcoxon_cutoff = lefse_wc, bootstrap_n = 30 , bootstrap_fraction = 2 / 3 , sample_min = 10 , multicls_strat = FALSE , curv = FALSE )<- purrr:: map_chr (i, ~ paste0 (., "_lefse_all" ))assign (tmp_name, tmp_lefse)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_lefse_asv" )objects (pattern = "_lefse_all" )``` ```{r} #| echo: false # DONT RUN, TESTING ONLY #tmp_tab <- data.frame(marker_table(ssu18_ps_pime_lefse_asv)) #tmp_tab$feature <- gsub('s__', '', tmp_tab$feature) #tmp_ps <- data.frame(t(otu_table(ssu18_ps_pime_rf_asv))) #tmp_ps <- tmp_ps %>% tibble::rownames_to_column("feature") #write.table(dplyr::left_join(tmp_tab, tmp_ps, by = "feature"), "test.txt", quote = FALSE, sep = "\t") ``` ```{r} #| code-fold: true for (i in samp_ps) {<- data.frame (tax_table (get (i)))$ ASV_ID <- NULL <- tmp_o_tax %>% tibble:: rownames_to_column ("ASV_ID" )<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_asv" )))<- data.frame (marker_table (tmp_get))$ feature <- gsub ('s__' , '' , tmp_mt$ feature)<- tmp_mt %>% dplyr:: rename (c ("ASV_ID" = 1 , "group" = 2 , "pval" = 4 )) %>% :: remove_rownames ()<- dplyr:: left_join (tmp_mt, tmp_o_tax, by = "ASV_ID" )$ group <- stringr:: str_replace (tmp_sum$ group, "^0$" , "C0" )$ group <- stringr:: str_replace (tmp_sum$ group, "^3$" , "W3" )$ group <- stringr:: str_replace (tmp_sum$ group, "^8$" , "W8" )<- tmp_sum[order (as.numeric (gsub ("[A-Z]{3}" , "" , tmp_sum$ ASV_ID))),]<- purrr:: map_chr (i, ~ paste0 (., "_lefse_summary" ))assign (tmp_res_name, tmp_sum)rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true for (i in samp_ps) {<- get (i)<- data.frame (t (otu_table (tmp_get)))#tmp_get[,1] <- NULL #tmp_df <- as.data.frame(t(tmp_otu)) <- colnames (tmp_df)<- tmp_col_names %>% :: str_replace ("P[0-9]{2}_D[0-9]{2}_[0-9]{3}_" , "" ) %>% :: str_replace ("[A-Z]$" , "" )colnames (tmp_df) <- tmp_col_names$ freq <- apply (tmp_df > 0 , 1 , sum)$ freq_C0 <- apply (tmp_df[ , (names (tmp_df) %in% "C0" )] > 0 , 1 , sum)$ freq_W3 <- apply (tmp_df[ , (names (tmp_df) %in% "W3" )] > 0 , 1 , sum)$ freq_W8 <- apply (tmp_df[ , (names (tmp_df) %in% "W8" )] > 0 , 1 , sum)$ reads_total <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% c ("C0" , "W3" , "W8" ))])$ reads_C0 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "C0" )])$ reads_W3 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "W3" )])$ reads_W8 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "W8" )])<- tmp_df[,! grepl ("^[C | W]" , names (tmp_df))]<- tmp_df %>% tibble:: rownames_to_column ("ASV_ID" )<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_summary" )))<- merge (tmp_df, tmp_get_lefse, by = "ASV_ID" , all = FALSE )<- tmp_merge_df[,c (1 ,10 : 12 ,2 : 9 ,14 : 20 )]<- tmp_merge_df[order (tmp_merge_df$ reads_total, decreasing = TRUE ), ]<- purrr:: map_chr (i, ~ paste0 (., "_lefse_final" ))assign (tmp_merge_name, tmp_merge_df)rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true for (i in samp_ps) {<- get (i)<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_summary" )))<- tmp_tab[,1 ]<- prune_taxa (tmp_list, tmp_get)<- purrr:: map_chr (i, ~ paste0 (., "_lefse" ))assign (tmp_name, tmp_ps)@ phy_tree <- NULL <- prune_samples (sample_sums (tmp_ps) > 0 , tmp_ps)<- rtree (ntaxa (tmp_ps), rooted = TRUE ,tip.label = taxa_names (tmp_ps))<- merge_phyloseq (tmp_ps, sample_data, tmp_ps_tree)assign (tmp_name, tmp_ps)rm (list = ls (pattern = "tmp_" ))objects ()``` ## LEfSe Summaries (ASVs & Markers) ### ASV summary data < br /> #### FULL ```{r} #| echo: false #| eval: true <- ssu18_ps_work_lefse_finalwrite.table (seq_table, "files/da/tables/ssu_lefse_results_full.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_lefse_full")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),ef_lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "ef_lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### Arbitrary filtered ```{r} #| echo: false #| eval: true <- ssu18_ps_filt_lefse_finalwrite.table (seq_table, "files/da/tables/ssu_lefse_results_arbitrary.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_lefse_filt")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PERfect filtered ```{r} #| echo: false #| eval: true <- ssu18_ps_perfect_lefse_finalwrite.table (seq_table, "files/da/tables/ssu_lefse_results_perfect.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_lefse_perfect")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PIME filtered ```{r} #| echo: false #| eval: true <- ssu18_ps_pime_lefse_finalwrite.table (seq_table, "files/da/tables/ssu_lefse_results_pime.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_lefse_pime")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` ### ASV Visualizations ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_asv" )))<- data.frame (marker_table (tmp_get))<- tmp_mt %>% filter (ef_lda >= 3 )<- marker_table (tmp_mt)<- purrr:: map_chr (i, ~ paste0 (., "_lefse_asv_viz_trim" ))assign (tmp_res_name, tmp_mt)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_lefse_asv_viz_trim" )``` </ br > #### FULL ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (ssu18_ps_work_lefse_asv_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_ssu("ssu_lefse_asv_full")` </ small > #### Arbitrary filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (ssu18_ps_filt_lefse_asv_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_ssu("ssu_lefse_asv_filt")` </ small > #### PERfect filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (ssu18_ps_perfect_lefse_asv_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 6 ))``` < small > `r caption_fig_ssu("ssu_lefse_asv_perfect")` </ small > #### PIME filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (ssu18_ps_pime_lefse_asv_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 5 ))``` < small > `r caption_fig_ssu("ssu_lefse_asv_pime")` </ small > ### Marker summary data ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_all" )))<- data.frame (marker_table (tmp_get))<- tmp_mt[, c (2 : 4 ,1 )]<- tmp_mt %>% dplyr:: rename (c ("group" = 1 , "pval" = 3 ))$ group <- stringr:: str_replace (tmp_mt$ group, "^0$" , "C0" )$ group <- stringr:: str_replace (tmp_mt$ group, "^3$" , "W3" )$ group <- stringr:: str_replace (tmp_mt$ group, "^8$" , "W8" )<- tmp_mt %>% tibble:: rownames_to_column ("marker" )<- tmp_mt %>% tidyr:: separate (col = "feature" , into = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" , "ASV_ID" ), sep = " \\ | \\ w__" )$ Kingdom <- gsub ('k__' , '' , tmp_mt$ Kingdom)<- purrr:: map_chr (i, ~ paste0 (., "_lefse_marker_final" ))assign (tmp_res_name, tmp_mt)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_lefse_marker_final" )``` </ br > #### FULL ```{r} #| echo: false #| eval: true <- ssu18_ps_work_lefse_marker_finalwrite.table (seq_table, "files/da/tables/ssu_lefse_marker_results_full.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_lefse_marker_full")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (marker = colDef (name = "Marker" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),ef_lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_ID = colDef (name = "ASV ID" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "ef_lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### Arbitrary filtered ```{r} #| echo: false #| eval: true <- ssu18_ps_filt_lefse_marker_finalwrite.table (seq_table, "files/da/tables/ssu_lefse_marker_results_arbitrary.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_lefse_marker_filt")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (marker = colDef (name = "Marker" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_ID = colDef (name = "ASV ID" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PERfect filtered ```{r} #| echo: false #| eval: true <- ssu18_ps_perfect_lefse_marker_finalwrite.table (seq_table, "files/da/tables/ssu_lefse_marker_results_perfect.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_lefse_marker_perfect")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (marker = colDef (name = "Marker" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_ID = colDef (name = "ASV ID" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PIME filtered ```{r} #| echo: false #| eval: true <- ssu18_ps_pime_lefse_marker_finalwrite.table (seq_table, "files/da/tables/ssu_lefse_marker_results_pime.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_ssu("ssu_lefse_marker_pime")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (marker = colDef (name = "Marker" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_ID = colDef (name = "ASV ID" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` ### Marker visualizations ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_all" )))<- data.frame (marker_table (tmp_get))<- tmp_mt %>% filter (ef_lda >= 4 )<- marker_table (tmp_mt)<- purrr:: map_chr (i, ~ paste0 (., "_lefse_all_viz_trim" ))assign (tmp_res_name, tmp_mt)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_lefse_all_viz_trim" )``` < br /> #### FULL ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (ssu18_ps_work_lefse_all_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_ssu("ssu_lefse_marker_full")` </ small > #### Arbitrary filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (ssu18_ps_filt_lefse_all_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_ssu("ssu_lefse_marker_filt")` </ small > #### PERfect filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (ssu18_ps_perfect_lefse_all_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_ssu("ssu_lefse_marker_perfect")` </ small > #### PIME filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (ssu18_ps_pime_lefse_all_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_ssu("ssu_lefse_marker_pime")` </ small > ## Response of select taxa [ `microbiomeMarker` ](https://github.com/yiluheihei/microbiomeMarker) package to assess the response of taxonomic lineages to soil warming. In the first step we need to fix the selected data set to make it compatible with the various functions. For this analysis we use the PERfect filtered data set.```{r} #| echo: false #| eval: false #TEMP LOAD ONLY REMOVE WHEN WORKFLOW FINISHED remove (list = ls ())load ("page_build/da_wf.rdata" ):: keep ("ssu18_ps_work_rf_all" , "ssu18_ps_work_rf_asv" , "ssu18_ps_filt_rf_all" , "ssu18_ps_filt_rf_asv" , "ssu18_ps_perfect_rf_all" , "ssu18_ps_perfect_rf_asv" , "ssu18_ps_pime_rf_all" , "ssu18_ps_pime_rf_asv" , sure = TRUE )objects ()``` ```{r} #| code-fold: true ## FIX ps object <- ssu18_ps_perfect_rf_all<- data.frame (tax_table (ssu_ps))#tmp_tax1$ASV_SEQ <- NULL <- row.names (tmp_tax1)<- data.frame (lapply (tmp_tax1, function (x) {gsub (" \\ (|)" , "" , x)}))row.names (tmp_tax) <- tmp_rnidentical (row.names (tmp_tax), row.names (tmp_tax1))#dplyr::filter(tmp_tax, Phylum == "Acidobacteriota") <- as.matrix (tmp_tax)<- phyloseq (otu_table (ssu_ps),phy_tree (ssu_ps),tax_table (ps_tax_new),sample_data (ssu_ps))<- tmp_ps:: rank_names (ssu_ps)data.frame (tax_table (ssu_ps))``` `run_test_multiple_groups` function.```{r} #| code-fold: true <- run_test_multiple_groups (ssu_ps, group = "TEMP" , taxa_rank = "all" , method = "anova" )@ marker_tablemarker_table (ssu_group_anova)``` ```{r} #| echo: false ## Loop through different norms #norm: #default = TSS #CSS = NONE = RLE = TMM #meth <- c("none", "rarefy", "TSS", "CLR") #for (i in meth) { # tmp_pht <- run_posthoc_test(ssu_ps, group = "TEMP", method = "tukey", norm = i, transform = "log10") # tmp_name <- purrr::map_chr(i, ~ paste0(., "_results")) # assign(tmp_name, tmp_pht) # rm(list = ls(pattern = "tmp_")) #} ## Filter results #filter(data.frame(CLR_results@result), group_name == "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales") #filter(data.frame(CSS_results@result), group_name == "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales") #filter(data.frame(none_results@result), group_name == "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales") #filter(data.frame(rarefy_results@result), group_name == "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales") #filter(data.frame(RLE_results@result), group_name == "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales") #filter(data.frame(TMM_results@result), group_name == "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales") ``` `run_posthoc_test` function.```{r} #| code-fold: true <- run_posthoc_test (ssu_ps, group = "TEMP" , method = "tukey" , transform = "log10" )``` ```{r} #| echo: true #| eval: true #filter(data.frame(ssu_default_pht@result), group_name == "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales") plot_postHocTest (ssu_default_pht, feature = "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales" ) & theme_bw ()``` ```{r} #| code-fold: true <- ssu_default_pht<- filter (data.frame (ssu_pht@ result), <= "0.05" )[! grepl ("ASV" , filter (data.frame (ssu_pht@ result), <= "0.05" )$ group_name),]<- ssu18_pht_filt[! grepl ("[a-z]__$" , ssu18_pht_filt$ group_name),]<- distinct (ssu18_pht_filt, group_name, .keep_all = TRUE )nrow (ssu18_pht_filt)save.image ("page_build/mmarker_wf.rdata" )``` < details > < summary > **Click here** to see a list of significantly different lineage markers.</ summary > ```{r} #| echo: false #| eval: true <- filter (data.frame (ssu_pht@ result), <= "0.05" )[! grepl ("ASV" , filter (data.frame (ssu_pht@ result), <= "0.05" )$ group_name),]<- seq_table[! grepl ("[a-z]__$" , seq_table$ group_name),]$ group <- NULL reactable (seq_table, rownames = FALSE ,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (group_name = colDef (name = "Lineage" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 150 ), comparions = colDef (name = "comparions" , align = "center" ),diff_mean = colDef (name = "diff_mean" , align = "center" , filterable = FALSE ,minWidth = 130 ),pvalue = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ci_lower = colDef (name = "ci_lower" , align = "center" , filterable = FALSE ),ci_upper = colDef (name = "ci_upper" , align = "center" , filterable = FALSE )searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` </ details > ```{r} #| echo: false source ("hack_code/plot_postHocTest_jjs.R" )environment (plot_postHocTest_jjs) <- asNamespace ('microbiomeMarker' )``` `r nrow(ssu18_pht_filt)` markers to plot and visualize the results.```{r} #| code-fold: true <- c ("k__Bacteria|p__Acidobacteriota|c__Acidobacteriae|o__Subgroup_2" , "k__Bacteria|p__Bacteroidota|c__Bacteroidia|o__Chitinophagales|f__Saprospiraceae" , "k__Bacteria|p__Bacteroidota|c__Bacteroidia|o__Cytophagales" , "k__Bacteria|p__Bacteroidota|c__Bacteroidia|o__Flavobacteriales" , "k__Bacteria|p__Bacteroidota|c__Bacteroidia|o__Sphingobacteriales" , "k__Bacteria|p__Myxococcota|c__Polyangia|o__mle1-27" , "k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Burkholderiales|f__Comamonadaceae|g__Rubrivivax" , "k__Bacteria|p__Actinobacteriota|c__Acidimicrobiia|o__Microtrichales" , "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales" , "k__Bacteria|p__Firmicutes|c__Bacilli|o__Bacillales" , "k__Bacteria|p__Myxococcota|c__Myxococcia|o__Myxococcales|f__Myxococcaceae|g__Corallococcus" , "k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Burkholderiales|f__Burkholderiaceae|g__Ralstonia" ``` ```{r} #| echo: false <- c ("#2271B2" , "#71B222" , "#B22271" )for (i in ssu_select) {<- i<- plot_postHocTest_jjs (ssu_pht, feature = tmp_select_feat) & theme_bw () <- tmp_plot + geom_boxplot (fill = swel_col) + scale_colour_manual (values = c ("#191919" , "#191919" , "#191919" )) + geom_point (size = 2 , show.legend = FALSE ) + ylab ("Relative abundance (% total reads)" )<- purrr:: map_chr (i, ~ paste0 (., "_tax_plot" ))#tmp_name <- purrr::map_chr(gsub(pattern = "^.*[a-z]\\|", x = i, replacement = ""), ~ paste0(., "_tax_plot")) assign (tmp_name, tmp_plot):: ggsave (tmp_plot, file = paste0 ("files/da/figures/" , i,".png" , sep = "" ), height = 7 , width = 9 , units = 'cm' , dpi = 600 , bg = "white" ):: ggsave (tmp_plot, file = paste0 ("files/da/figures/" , i,".pdf" , sep = "" ), height = 5 , width = 6.5 )rm (list = ls (pattern = "tmp_" ))``` ```{r} #| echo: false <- c ("Subgroup_2 (Acidobacteriota)" , "Saprospiraceae (Bacteroidota)" , "Cytophagales (Bacteroidota)" , "Flavobacteriales (Bacteroidota)" , "Sphingobacteriales (Bacteroidota)" , "mle1-27 (Myxococcota)" , "Rubrivivax (Proteobacteria)" , "Microtrichales (Actinobacteriota)" , "Gaiellales (Actinobacteriota)" , "Bacillales (Firmicutes)" , "Corallococcus (Myxococcota)" , "Ralstonia (Proteobacteria)" ``` ```{r} #| echo: false <- get (purrr:: map_chr (ssu_select[1 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[1 ])<- get (purrr:: map_chr (ssu_select[2 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[2 ])<- get (purrr:: map_chr (ssu_select[3 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[3 ])<- get (purrr:: map_chr (ssu_select[4 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[4 ])<- get (purrr:: map_chr (ssu_select[5 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[5 ])<- get (purrr:: map_chr (ssu_select[6 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[6 ])<- get (purrr:: map_chr (ssu_select[7 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[7 ])<- get (purrr:: map_chr (ssu_select[8 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[8 ])<- get (purrr:: map_chr (ssu_select[9 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[9 ])<- get (purrr:: map_chr (ssu_select[10 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[10 ])<- get (purrr:: map_chr (ssu_select[11 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[11 ])<- get (purrr:: map_chr (ssu_select[12 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (ssu_title[12 ])<- ((tmp_1 + tmp_2 + tmp_3) / (tmp_4 + tmp_5 + tmp_6) / (tmp_7 + tmp_8 + tmp_9) / (tmp_10 + tmp_11 + tmp_12))``` ```{r} #| echo: false :: ggsave (ssu_taxa_combo_plot, path = "files/da/figures/combo_plots" , filename = "ssu_taxa_marker.png" , height = 32 , width = 36 ,units = 'cm' , dpi = 600 , bg = "white" ):: ggsave (ssu_taxa_combo_plot, path = "files/da/figures/combo_plots" , filename = "ssu_taxa_marker.pdf" , height = 12 , width = 12 )``` ```{r} #| echo: false system ("cp files/da/figures/combo_plots/ssu_taxa_marker.png include/da/ssu_taxa_marker.png" )``` ```{r} #| echo: false #| eval: true #| warning: false #| column: body-outset :: include_graphics ("include/da/ssu_taxa_marker.png" )``` < small > `r caption_fig_ssu("ssu_taxa_marker_plot")` </ small > ```{r} #| echo: false save.image ("page_build/mmarker_wf.rdata" )``` # ITS ```{r} #| include: false ## Initial Load for ANALYSIS #1 remove (list = ls ())<- readRDS ("files/data-prep/rdata/its18_ps_work.rds" )<- readRDS ("files/filtering/arbitrary/rdata/its18_ps_filt.rds" )<- readRDS ("files/filtering/perfect/rdata/its18_ps_perfect.rds" )<- readRDS ("files/filtering/pime/rdata/its18_ps_pime.rds" )``` ```{r} #| echo: false <- c ("its18_ps_work" , "its18_ps_filt" , "its18_ps_perfect" , "its18_ps_pime" ) for (i in samp_ps) { <- get (i) <- file.path ("files/alpha/rdata/" ) <- readRDS (paste (tmp_path, i, ".rds" , sep = "" )) <- sample_data (tmp_read) <- data.frame (tax_table (tmp_get)) #tmp_tax_data[,c(1:6)] $ ASV_ID <- NULL # Some have, some do not <- data.frame (lapply (tmp_tax_data, gsub, pattern = "^[k | p | c | o | f]_.*" , replacement = NA , fixed = FALSE )) <- as.matrix (tmp_tax_data) <- phyloseq (otu_table (tmp_get), tax_table (tmp_tax_data), assign (i, tmp_ps) rm (list = ls (pattern = "tmp_" )) objects () ``` ```{r} #| echo: false <- c ("#2271B2" , "#71B222" , "#B22271" )``` ## Indicator Analysis 1. Choose a data set(s) & set the p-value cutoff.```{r} <- c ("its18_ps_work" , "its18_ps_filt" , "its18_ps_perfect" , "its18_ps_pime" ) <- 0.05 ``` 2. Format Data Frame```{r} #| code-fold: true for (i in samp_ps) {<- get (i)<- data.frame (otu_table (tmp_get))<- tmp_df[, which (colSums (tmp_df) != 0 )]<- row.names (tmp_df)<- tmp_row_names %>% :: str_replace ("P[0-9]{2}_D[0-9]{2}_[0-9]{3}_" , "" ) %>% :: str_replace ("[A-Z]$" , "" )<- tmp_df %>% tibble:: add_column (tmp_row_names, .before = 1 )<- purrr:: map_chr (i, ~ paste0 (., "_seq_tab" ))assign (tmp_name, tmp_df)rm (list = ls (pattern = "tmp_" ))objects ()``` 3. Run Indicator Analysis```{r} #| code-fold: true set.seed (1191 )for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_seq_tab" )))<- indval (tmp_get[,- 1 ], tmp_get[,1 ])<- tmp_iva$ maxcls[tmp_iva$ pval <= indval_pval]<- tmp_iva$ indcls[tmp_iva$ pval <= indval_pval]<- tmp_iva$ pval[tmp_iva$ pval <= indval_pval]<- apply (tmp_get[,- 1 ] > 0 , 2 , sum)[tmp_iva$ pval <= indval_pval]<- data.frame (group = tmp_gr, indval = tmp_iv,pval = tmp_pv, freq = tmp_fr)<- tmp_sum[order (tmp_sum$ group, - tmp_sum$ indval),]<- data.frame (tax_table (get (i)))$ ASV_ID <- NULL <- merge (tmp_sum, tmp_tax_df, by = "row.names" , all = TRUE )<- tmp_sum_tax[! (is.na (tmp_sum_tax$ group)),]class (tmp_sum_tax$ group) <- "character" $ group <- stringr:: str_replace (tmp_sum_tax$ group, "^1$" , "C0" )$ group <- stringr:: str_replace (tmp_sum_tax$ group, "^2$" , "W3" )$ group <- stringr:: str_replace (tmp_sum_tax$ group, "^3$" , "W8" )<- tmp_sum_tax %>% dplyr:: rename ("ASV_ID" = "Row.names" )<- tmp_sum_tax[order (as.numeric (gsub ("[A-Z]{3}" , "" , tmp_sum_tax$ ASV_ID))),]$ ASV_ID <- as.character (tmp_sum_tax$ ASV_ID)<- p.adjust (tmp_sum$ pval, "bonferroni" )<- purrr:: map_chr (i, ~ paste0 (., "_indval_summary" ))assign (tmp_res_name, tmp_sum_tax)rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_seq_tab" )))1 ] <- NULL <- as.data.frame (t (tmp_get))<- colnames (tmp_df)<- tmp_col_names %>% :: str_replace ("P[0-9]{2}_D[0-9]{2}_[0-9]{3}_" , "" ) %>% :: str_replace ("[A-Z]$" , "" )colnames (tmp_df) <- tmp_col_names#tmp_df$freq_all <- apply(tmp_df > 0, 1, sum) $ freq_C0 <- apply (tmp_df[ , (names (tmp_df) %in% "C0" )] > 0 , 1 , sum)$ freq_W3 <- apply (tmp_df[ , (names (tmp_df) %in% "W3" )] > 0 , 1 , sum)$ freq_W8 <- apply (tmp_df[ , (names (tmp_df) %in% "W8" )] > 0 , 1 , sum)$ reads_total <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% c ("C0" , "W3" , "W8" ))])$ reads_C0 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "C0" )])$ reads_W3 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "W3" )])$ reads_W8 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "W8" )])<- tmp_df[,! grepl ("^[C | W]" , names (tmp_df))]<- tmp_df %>% tibble:: rownames_to_column ("ASV_ID" )<- get (purrr:: map_chr (i, ~ paste0 (., "_indval_summary" )))<- merge (tmp_df, tmp_get_indval, by = "ASV_ID" , all = FALSE )<- tmp_merge_df[,c (1 ,9 : 12 ,2 : 8 ,13 : 19 )]<- tmp_merge_df[order (tmp_merge_df$ reads_total, decreasing = TRUE ), ]<- purrr:: map_chr (i, ~ paste0 (., "_indval_final" ))assign (tmp_merge_name, tmp_merge_df)rm (list = ls (pattern = "tmp_" ))``` 4. Save a new phyloseq object```{r} #| code-fold: true for (i in samp_ps) {<- get (i)<- get (purrr:: map_chr (i, ~ paste0 (., "_indval_summary" )))<- tmp_tab[,1 ]<- prune_taxa (tmp_list, tmp_get)<- purrr:: map_chr (i, ~ paste0 (., "_ind" ))assign (tmp_name, tmp_ps)@ phy_tree <- NULL <- prune_samples (sample_sums (tmp_ps) > 0 , tmp_ps)<- rtree (ntaxa (tmp_ps), rooted = TRUE ,tip.label = taxa_names (tmp_ps))<- merge_phyloseq (tmp_ps, sample_data, tmp_ps_tree)assign (tmp_name, tmp_ps)rm (list = ls (pattern = "tmp_" ))objects ()``` ## Indicator Summary </ br > #### FULL ```{r} #| echo: false #| eval: true <- its18_ps_work_indval_finalwrite.table (seq_table, "files/da/tables/its_isa_results_full.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_isa_full")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),indval = colDef (name = "indval score" , align = "center" , filterable = FALSE ,minWidth = 130 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "Indicator analysis results" , sticky = "left" ,columns = c ("group" , "indval" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### Arbitrary filtered ```{r} #| echo: false #| eval: true <- its18_ps_filt_indval_finalwrite.table (seq_table, "files/da/tables/its_isa_results_arbitrary.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_isa_filt")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),indval = colDef (name = "indval score" , align = "center" , filterable = FALSE ,minWidth = 130 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "Indicator analysis results" , sticky = "left" ,columns = c ("group" , "indval" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PERfect filtered ```{r} #| echo: false #| eval: true <- its18_ps_perfect_indval_finalwrite.table (seq_table, "files/da/tables/its_isa_results_perfect.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_isa_perfect")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),indval = colDef (name = "indval score" , align = "center" , filterable = FALSE ,minWidth = 130 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "Indicator analysis results" , sticky = "left" ,columns = c ("group" , "indval" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PIME filtered ```{r} #| echo: false #| eval: true <- its18_ps_pime_indval_finalwrite.table (seq_table, "files/da/tables/its_isa_results_pime.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_isa_pime")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),indval = colDef (name = "indval score" , align = "center" , filterable = FALSE ,minWidth = 130 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "Indicator analysis results" , sticky = "left" ,columns = c ("group" , "indval" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` ## LEfSe Analysis [ microbiomeMarker ](https://github.com/yiluheihei/microbiomeMarker) ```{r} #| code-fold: true for (i in samp_ps) {<- get (i)tax_table (tmp_get) <- tax_table (tmp_get)[,c (1 : 6 )]tax_table (tmp_get) <- cbind (tax_table (tmp_get),rownames (tax_table (tmp_get)))colnames (tax_table (tmp_get)) <- c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" , "Species" )<- purrr:: map_chr (i, ~ paste0 (., "_rf_all" ))assign (tmp_name, tmp_get)tax_table (tmp_get) <- tax_table (tmp_get)[,c (7 )]<- purrr:: map_chr (i, ~ paste0 (., "_rf_asv" ))assign (tmp_name_asv, tmp_get)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_rf" )``` 1. Choose a data set(s) & set the LEfSe cutoff values. ```{r} <- c ("its18_ps_work" , "its18_ps_filt" , "its18_ps_perfect" , "its18_ps_pime" ) <- 2 <- 0.05 <- 0.05 ``` ```{r} #| warnings: false #| error: false #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_rf_asv" )))<- run_lefse (tmp_get, norm = "CPM" , class = "TEMP" , correct = "2" , lda_cutoff = lefse_lda, kw_cutoff = lefse_kw, wilcoxon_cutoff = lefse_wc, bootstrap_n = 30 , bootstrap_fraction = 2 / 3 , sample_min = 10 , multicls_strat = FALSE , curv = FALSE )<- purrr:: map_chr (i, ~ paste0 (., "_lefse_asv" ))assign (tmp_name, tmp_lefse)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_lefse_asv" )for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_rf_all" )))<- run_lefse (tmp_get, norm = "CPM" , class = "TEMP" , correct = "2" , lda_cutoff = lefse_lda, kw_cutoff = lefse_kw, wilcoxon_cutoff = lefse_wc, bootstrap_n = 30 , bootstrap_fraction = 2 / 3 , sample_min = 10 , multicls_strat = FALSE , curv = FALSE )<- purrr:: map_chr (i, ~ paste0 (., "_lefse_all" ))assign (tmp_name, tmp_lefse)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_lefse_asv" )objects (pattern = "_lefse_all" )``` ```{r} #| echo: false # DONT RUN, TESTING ONLY #tmp_tab <- data.frame(marker_table(its18_ps_pime_lefse_asv)) #tmp_tab$feature <- gsub('s__', '', tmp_tab$feature) #tmp_ps <- data.frame(t(otu_table(its18_ps_pime_rf_asv))) #tmp_ps <- tmp_ps %>% tibble::rownames_to_column("feature") #write.table(dplyr::left_join(tmp_tab, tmp_ps, by = "feature"), "test.txt", quote = FALSE, sep = "\t") ``` ```{r} #| code-fold: true for (i in samp_ps) {<- data.frame (tax_table (get (i)))$ ASV_ID <- NULL <- tmp_o_tax %>% tibble:: rownames_to_column ("ASV_ID" )<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_asv" )))<- data.frame (marker_table (tmp_get))$ feature <- gsub ('s__' , '' , tmp_mt$ feature)<- tmp_mt %>% dplyr:: rename (c ("ASV_ID" = 1 , "group" = 2 , "pval" = 4 )) %>% :: remove_rownames ()<- dplyr:: left_join (tmp_mt, tmp_o_tax, by = "ASV_ID" )$ group <- stringr:: str_replace (tmp_sum$ group, "^0$" , "C0" )$ group <- stringr:: str_replace (tmp_sum$ group, "^3$" , "W3" )$ group <- stringr:: str_replace (tmp_sum$ group, "^8$" , "W8" )<- tmp_sum[order (as.numeric (gsub ("[A-Z]{3}" , "" , tmp_sum$ ASV_ID))),]<- purrr:: map_chr (i, ~ paste0 (., "_lefse_summary" ))assign (tmp_res_name, tmp_sum)rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true for (i in samp_ps) {<- get (i)<- data.frame (t (otu_table (tmp_get)))#tmp_get[,1] <- NULL #tmp_df <- as.data.frame(t(tmp_otu)) <- colnames (tmp_df)<- tmp_col_names %>% :: str_replace ("P[0-9]{2}_D[0-9]{2}_[0-9]{3}_" , "" ) %>% :: str_replace ("[A-Z]$" , "" )colnames (tmp_df) <- tmp_col_names$ freq <- apply (tmp_df > 0 , 1 , sum)$ freq_C0 <- apply (tmp_df[ , (names (tmp_df) %in% "C0" )] > 0 , 1 , sum)$ freq_W3 <- apply (tmp_df[ , (names (tmp_df) %in% "W3" )] > 0 , 1 , sum)$ freq_W8 <- apply (tmp_df[ , (names (tmp_df) %in% "W8" )] > 0 , 1 , sum)$ reads_total <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% c ("C0" , "W3" , "W8" ))])$ reads_C0 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "C0" )])$ reads_W3 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "W3" )])$ reads_W8 <- base:: rowSums (tmp_df[ , (names (tmp_df) %in% "W8" )])<- tmp_df[,! grepl ("^[C | W]" , names (tmp_df))]<- tmp_df %>% tibble:: rownames_to_column ("ASV_ID" )<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_summary" )))<- merge (tmp_df, tmp_get_lefse, by = "ASV_ID" , all = FALSE )<- tmp_merge_df[,c (1 ,10 : 12 ,2 : 9 ,14 : 20 )]<- tmp_merge_df[order (tmp_merge_df$ reads_total, decreasing = TRUE ), ]<- purrr:: map_chr (i, ~ paste0 (., "_lefse_final" ))assign (tmp_merge_name, tmp_merge_df)rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true for (i in samp_ps) {<- get (i)<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_summary" )))<- tmp_tab[,1 ]<- prune_taxa (tmp_list, tmp_get)<- purrr:: map_chr (i, ~ paste0 (., "_lefse" ))assign (tmp_name, tmp_ps)@ phy_tree <- NULL <- prune_samples (sample_sums (tmp_ps) > 0 , tmp_ps)<- rtree (ntaxa (tmp_ps), rooted = TRUE ,tip.label = taxa_names (tmp_ps))<- merge_phyloseq (tmp_ps, sample_data, tmp_ps_tree)assign (tmp_name, tmp_ps)rm (list = ls (pattern = "tmp_" ))objects ()``` ## LEfSe Summaries (ASVs & Markers) ### ASV summary data < br /> #### FULL ```{r} #| echo: false #| eval: true <- its18_ps_work_lefse_finalwrite.table (seq_table, "files/da/tables/its_lefse_results_full.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_lefse_full")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),ef_lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "ef_lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### Arbitrary filtered ```{r} #| echo: false #| eval: true <- its18_ps_filt_lefse_finalwrite.table (seq_table, "files/da/tables/its_lefse_results_arbitrary.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_lefse_filt")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PERfect filtered ```{r} #| echo: false #| eval: true <- its18_ps_perfect_lefse_finalwrite.table (seq_table, "files/da/tables/its_lefse_results_perfect.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_lefse_perfect")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PIME filtered ```{r} #| echo: false #| eval: true <- its18_ps_pime_lefse_finalwrite.table (seq_table, "files/da/tables/its_lefse_results_pime.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_lefse_pime")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (ASV_ID = colDef (name = "ASV_ID" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),freq = colDef (name = "all samples" , align = "center" , filterable = FALSE ),freq_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),freq_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),freq_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),reads_total = colDef (name = "all" , align = "center" , filterable = FALSE ),reads_C0 = colDef (name = "control" , align = "center" , filterable = FALSE ),reads_W3 = colDef (name = "+3°C" , align = "center" , filterable = FALSE ),reads_W8 = colDef (name = "+8°C" , align = "center" , filterable = FALSE , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_SEQ = colDef (name = "ASV sequence" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Frequency" , columns = c ("freq" , "freq_C0" , "freq_W3" , "freq_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Read totals" , columns = c ("reads_total" , "reads_C0" , "reads_W3" , "reads_W8" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` ### ASV Visualizations ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_asv" )))<- data.frame (marker_table (tmp_get))<- tmp_mt %>% filter (ef_lda >= 3 )<- marker_table (tmp_mt)<- purrr:: map_chr (i, ~ paste0 (., "_lefse_asv_viz_trim" ))assign (tmp_res_name, tmp_mt)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_lefse_asv_viz_trim" )``` </ br > #### FULL ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (its18_ps_work_lefse_asv_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_its("its_lefse_asv_full")` </ small > #### Arbitrary filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (its18_ps_filt_lefse_asv_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_its("its_lefse_asv_filt")` </ small > #### PERfect filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (its18_ps_perfect_lefse_asv_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 6 ))``` < small > `r caption_fig_its("its_lefse_asv_perfect")` </ small > #### PIME filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (its18_ps_pime_lefse_asv_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 5 ))``` < small > `r caption_fig_its("its_lefse_asv_pime")` </ small > ### Marker summary data ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_all" )))<- data.frame (marker_table (tmp_get))<- tmp_mt[, c (2 : 4 ,1 )]<- tmp_mt %>% dplyr:: rename (c ("group" = 1 , "pval" = 3 ))$ group <- stringr:: str_replace (tmp_mt$ group, "^0$" , "C0" )$ group <- stringr:: str_replace (tmp_mt$ group, "^3$" , "W3" )$ group <- stringr:: str_replace (tmp_mt$ group, "^8$" , "W8" )<- tmp_mt %>% tibble:: rownames_to_column ("marker" )<- tmp_mt %>% tidyr:: separate (col = "feature" , into = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" , "ASV_ID" ), sep = " \\ | \\ w__" )$ Kingdom <- gsub ('k__' , '' , tmp_mt$ Kingdom)<- purrr:: map_chr (i, ~ paste0 (., "_lefse_marker_final" ))assign (tmp_res_name, tmp_mt)rm (list = ls (pattern = "tmp_" ))``` < br /> #### FULL ```{r} #| echo: false #| eval: true <- its18_ps_work_lefse_marker_finalwrite.table (seq_table, "files/da/tables/its_lefse_marker_results_full.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_lefse_marker_full")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (marker = colDef (name = "Marker" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),ef_lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_ID = colDef (name = "ASV ID" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "ef_lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### Arbitrary filtered ```{r} #| echo: false #| eval: true <- its18_ps_filt_lefse_marker_finalwrite.table (seq_table, "files/da/tables/its_lefse_marker_results_arbitrary.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_lefse_marker_filt")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (marker = colDef (name = "Marker" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_ID = colDef (name = "ASV ID" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PERfect filtered ```{r} #| echo: false #| eval: true <- its18_ps_perfect_lefse_marker_finalwrite.table (seq_table, "files/da/tables/its_lefse_marker_results_perfect.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_lefse_marker_perfect")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (marker = colDef (name = "Marker" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_ID = colDef (name = "ASV ID" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` #### PIME filtered ```{r} #| echo: false #| eval: true <- its18_ps_pime_lefse_marker_finalwrite.table (seq_table, "files/da/tables/its_lefse_marker_results_pime.txt" , row.names = FALSE , sep = " \t " , quote = FALSE )``` < small > `r caption_tab_its("its_lefse_marker_pime")` </ small > ```{r} #| echo: false #| eval: true reactable (seq_table,defaultColDef = colDef (headerStyle = list (fontSize = "0.8em" ),header = function (value) gsub ("_" , " " , value, fixed = TRUE ),cell = function (value) format (value, nsmall = 0 ),align = "left" , filterable = TRUE , sortable = TRUE , resizable = TRUE ,footerStyle = list (fontWeight = "bold" )columns = list (marker = colDef (name = "Marker" , sticky = "left" , style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" ), align = "left" ,minWidth = 100 ), group = colDef (name = "enriched" , align = "center" ),lda = colDef (name = "LDA score" , align = "center" , filterable = FALSE ,minWidth = 100 ),pval = colDef (name = "p-value" , align = "center" , filterable = FALSE ,style = list (borderRight = "5px solid #eee" ),headerStyle = list (borderRight = "5px solid #eee" , fontSize = "0.8em" )),ASV_ID = colDef (name = "ASV ID" , filterable = FALSE )columnGroups = list (colGroup (name = "LEfSe results" , sticky = "left" ,columns = c ("group" , "lda" , "pval" ), headerStyle = list (fontSize = "1.2em" , borderRight = "5px solid #eee" )),colGroup (name = "Lineage" , align = "left" ,columns = c ("Kingdom" , "Phylum" , "Class" , "Order" , "Family" , "Genus" ))searchable = TRUE , defaultPageSize = 5 , pageSizeOptions = c (5 , 10 , nrow (seq_table)), showPageSizeOptions = TRUE , highlight = TRUE , bordered = TRUE , striped = TRUE , compact = FALSE , wrap = FALSE , showSortable = TRUE , fullWidth = TRUE ,theme = reactableTheme (style = list (fontSize = "0.8em" )))rm (seq_table)``` ### Marker visualizations ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_all" )))<- data.frame (marker_table (tmp_get))<- tmp_mt %>% filter (ef_lda >= 3.5 )<- marker_table (tmp_mt)<- purrr:: map_chr (i, ~ paste0 (., "_lefse_all_viz_trim" ))assign (tmp_res_name, tmp_mt)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_lefse_all_viz_trim" )``` #### FULL ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (its18_ps_work_lefse_all_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_its("its_lefse_marker_full")` </ small > #### Arbitrary filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (its18_ps_filt_lefse_all_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_its("its_lefse_marker_filt")` </ small > #### PERfect filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (its18_ps_perfect_lefse_all_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_its("its_lefse_marker_perfect")` </ small > #### PIME filtered ```{r} #| echo: false #| eval: true #| warning: false plot_ef_bar (its18_ps_pime_lefse_all_viz_trim) + scale_fill_manual (values = c ("0" = "#2271B2" , "3" = "#71B222" , "8" = "#B22271" )) + theme (axis.text = element_text (size = 8 ))``` < small > `r caption_fig_its("its_lefse_marker_pime")` </ small > ## Response of select taxa < details > < summary > **Click here** to see the ITS version of this workflow.</ summary > ```{r} #| echo: false #| eval: false #TEMP LOAD ONLY REMOVE WHEN WORKFLOW FINISHED remove (list = ls ())load ("page_build/da_wf.rdata" ):: keep ("its18_ps_work_rf_all" , "its18_ps_work_rf_asv" , "its18_ps_filt_rf_all" , "its18_ps_filt_rf_asv" , "its18_ps_perfect_rf_all" , "its18_ps_perfect_rf_asv" , "its18_ps_pime_rf_all" , "its18_ps_pime_rf_asv" , sure = TRUE )objects ()``` ```{r} ## FIX ps object <- its18_ps_perfect_rf_all<- data.frame (tax_table (ps))$ ASV_SEQ <- NULL <- row.names (tmp_tax1)<- data.frame (lapply (tmp_tax1, function (x) {gsub (" \\ (|)" , "" , x)}))row.names (tmp_tax) <- tmp_rnidentical (row.names (tmp_tax), row.names (tmp_tax1))#dplyr::filter(tmp_tax, Phylum == "Acidobacteriota") <- as.matrix (tmp_tax)<- phyloseq (otu_table (ps),tax_table (ps_tax_new),sample_data (ps))<- tmp_ps:: rank_names (its_ps)data.frame (tax_table (its_ps))``` ```{r} <- run_test_multiple_groups (its_ps, group = "TEMP" , taxa_rank = "all" , method = "anova" )@ marker_tablemarker_table (its_group_anova)``` ```{r} <- run_posthoc_test (its_ps, group = "TEMP" , method = "tukey" , transform = "log10" )``` ```{r} #| echo: false #filter(data.frame(its_default_pht@result), group_name == "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales") #plot_postHocTest(its_default_pht, feature = "k__Bacteria|p__Actinobacteriota|c__Thermoleophilia|o__Gaiellales") & theme_bw() ``` ```{r} <- its_default_pht<- filter (data.frame (its_pht@ result), pvalue <= "0.05" )[! grepl ("ASV" , filter (data.frame (its_pht@ result), pvalue <= "0.05" )$ group_name),]<- its18_pht_filt[! grepl ("[a-z]__$" , its18_pht_filt$ group_name),]<- unique (its18_pht_filt$ group_name)length (its18_pht_filt)``` < details > < summary > **Click here** to see a list of significantly different lineage markers.</ summary > ```{r} #| echo: false #| eval: true ``` </ details > ```{r} #| echo: false source ("hack_code/plot_postHocTest_jjs.R" )environment (plot_postHocTest_jjs) <- asNamespace ('microbiomeMarker' )``` ```{r} #| echo: false <- c ("k__Fungi|p__Ascomycota|c__Sordariomycetes|o__Xylariales|f__Microdochiaceae" , "k__Fungi|p__Basidiomycota|c__Agaricomycetes|o__Agaricales|f__Entolomataceae" , "k__Fungi|p__Basidiomycota|c__Agaricomycetes|o__Agaricales|f__Clavariaceae" , "k__Fungi|p__Basidiomycota|c__Agaricomycetes|o__Agaricales" , "k__Fungi|p__Basidiomycota|c__Microbotryomycetes|o__Sporidiobolales" , "k__Fungi|p__Rozellomycota|c__Rozellomycotina_cls_Incertae_sedis" , "k__Fungi|p__Ascomycota|c__Eurotiomycetes|o__Eurotiales|f__Trichocomaceae|g__Talaromyces" , "k__Fungi|p__Ascomycota|c__Pezizomycetes|o__Pezizales|f__Pyronemataceae" , "k__Fungi|p__Ascomycota|c__Sordariomycetes|o__Hypocreales|f__Nectriaceae|g__Fusarium" , "k__Fungi|p__Ascomycota|c__Saccharomycetes|o__Saccharomycetales|f__Metschnikowiaceae" , "k__Fungi|p__Glomeromycota|c__Glomeromycetes|o__Glomerales" , "k__Fungi|p__Mortierellomycota|c__Mortierellomycetes|o__Mortierellales" ``` ```{r} #| echo: false <- c ("#2271B2" , "#71B222" , "#B22271" )for (i in its_select) {<- i<- plot_postHocTest_jjs (its_pht, feature = tmp_select_feat) & theme_bw () <- tmp_plot + geom_boxplot (fill = swel_col) + scale_colour_manual (values = c ("#191919" , "#191919" , "#191919" )) + geom_point (size = 2 , show.legend = FALSE ) + ylab ("Relative abundance (% total reads)" )<- purrr:: map_chr (i, ~ paste0 (., "_tax_plot" ))#tmp_name <- purrr::map_chr(gsub(pattern = "^.*[a-z]\\|", x = i, replacement = ""), ~ paste0(., "_tax_plot")) assign (tmp_name, tmp_plot):: ggsave (tmp_plot, file = paste0 ("files/da/figures/" , i,".png" , sep = "" ), height = 7 , width = 9 , units = 'cm' , dpi = 600 , bg = "white" ):: ggsave (tmp_plot, file = paste0 ("files/da/figures/" , i,".pdf" , sep = "" ), height = 5 , width = 6.5 )rm (list = ls (pattern = "tmp_" ))``` ```{r} #| echo: false <- c ("Microdochiaceae (Ascomycota)" , "Entolomataceae (Basidiomycota)" , "Clavariaceae (Basidiomycota)" , "Agaricales (Basidiomycota)" , "Sporidiobolales (Basidiomycota)" , "Rozellomycotina (Rozellomycota)" , "Talaromyces (Ascomycota)" , "Pyronemataceae (Ascomycota)" , "Fusarium (Ascomycota)" , "Metschnikowiaceae (Ascomycota)" , "Glomerales (Glomeromycota)" , "Mortierellales (Mortierellomycota)" ``` ```{r} #| echo: false <- get (purrr:: map_chr (its_select[1 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[1 ])<- get (purrr:: map_chr (its_select[2 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[2 ])<- get (purrr:: map_chr (its_select[3 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[3 ])<- get (purrr:: map_chr (its_select[4 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[4 ])<- get (purrr:: map_chr (its_select[5 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[5 ])<- get (purrr:: map_chr (its_select[6 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[6 ])<- get (purrr:: map_chr (its_select[7 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[7 ])<- get (purrr:: map_chr (its_select[8 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[8 ])<- get (purrr:: map_chr (its_select[9 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[9 ])<- get (purrr:: map_chr (its_select[10 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[10 ])<- get (purrr:: map_chr (its_select[11 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[11 ])<- get (purrr:: map_chr (its_select[12 ], ~ paste0 (., "_tax_plot" ))) + geom_point (show.legend = FALSE ) + ggtitle (its_title[12 ])<- ((tmp_1 + tmp_2 + tmp_3) / (tmp_4 + tmp_5 + tmp_6) / (tmp_7 + tmp_8 + tmp_9) / (tmp_10 + tmp_11 + tmp_12))``` ```{r} #| echo: false :: ggsave (its_taxa_combo_plot, path = "files/da/figures/combo_plots" , filename = "its_taxa_marker.png" , height = 32 , width = 36 ,units = 'cm' , dpi = 600 , bg = "white" ):: ggsave (its_taxa_combo_plot, path = "files/da/figures/combo_plots" , filename = "its_taxa_marker.pdf" , height = 12 , width = 12 )``` ```{r} #| echo: false system ("cp files/da/figures/combo_plots/its_taxa_marker.png include/da/its_taxa_marker.png" )``` </ details > ```{r} #| echo: false #| eval: true #| warning: false #| column: body-outset :: include_graphics ("include/da/ssu_taxa_marker.png" )``` < small > `r caption_fig_its("its_taxa_marker_plot")` </ small > # Visualizing DA ASVs in Anvi’o [ anvi’o ](https://github.com/merenlab/anvio) ---an advanced analysis and visualization platform for ‘omics data [ @eren2015anvi ] ---using the `anvi-interactive` command. Anvi’o likes databases but it also understands that sometimes you do not have a database. So it offers a manual mode. If you type this command you can have a look at the relevant pieces we need for the visualization, specifically those under the headings MANUAL INPUTS and ADDITIONAL STUFF.```bash anvi-interactive -h ``` ``` MANUAL INPUTS: Mandatory input parameters to start the interactive interface without anvi'o databases. --manual-mode We need this flag to run anvi'o in an ad hoc manner, i.e., no database. -f FASTA, --fasta-file FASTA A FASTA-formatted input file. This is sort of optional -d VIEW_DATA, --view-data VIEW_DATA A TAB-delimited file for view data. This is the ASV by sample matrix. We need this -t NEWICK, --tree NEWICK NEWICK formatted tree structure. How the ASVs are ordered in our case. ADDITIONAL STUFF: Parameters to provide additional layers, views, or layer data. -V ADDITIONAL_VIEW, --additional-view ADDITIONAL_VIEW A TAB-delimited file for an additional view to be used in the interface. This file should contain all split names, and values for each of them in all samples. Each column in this file must correspond to a sample name. Content of this file will be called 'user_view', which will be available as a new item in the 'views' combo box in the interface -A ADDITIONAL_LAYERS, --additional-layers ADDITIONAL_LAYERS A TAB-delimited file for additional layer info. In our case this is info about each ASV. The first column of the file must be the ASV names, and the remaining columns should be unique attributes. ``` [ Working with anvi’o additional data tables ](http://merenlab.org/2017/12/11/additional-data-tables/) . A lot of what we need is covered in this tutorial. To get the most out the visualization we need to create a few files to give anvi’o when we fire up the interactive interface.1. View data: in our case, a sample by ASV abundance matrix.2. Additional info about each ASV.3. Additional info about each sample.4. Taxa abundance data for each sample at some rank.5. Dendrograms ordering the ASVs and samples (based on view data).6. Fasta file of all ASVs in the analysis.## Main Steps ### 1. View data `-d` or `--view-data` file. This file needs to be an ASV by sample matrix of read counts. To simplify the visualization, we will use ***all*** ASVs represented by 100 or more total reads, including those identified as differentially abundant by the ISA and/or LEfSe. ```{r} #| code-fold: true <- c ("ssu18_ps_filt" , "ssu18_ps_pime" , "ssu18_ps_perfect" )<- c ("ssu18_ps_filt" , "ssu18_ps_pime" , "ssu18_ps_perfect" )<- c ("ssu18_ps_work" )<- c ("ssu18_ps_pime" )<- c ("ssu18_ps_filt" , "ssu18_ps_perfect" )<- 100 for (i in samp_ps_pime) {<- get (i)<- prune_taxa (taxa_sums (tmp_get) > trim_val, tmp_get)<- purrr:: map_chr (i, ~ paste0 (., "_trim" ))assign (tmp_name, tmp_df)rm (list = ls (pattern = "tmp_" ))<- 300 for (i in samp_ps_other) {<- get (i)<- prune_taxa (taxa_sums (tmp_get) > trim_val, tmp_get)<- purrr:: map_chr (i, ~ paste0 (., "_trim" ))assign (tmp_name, tmp_df)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_trim" )``` ```{r} #| echo: false for (i in samp_ps) {<- file.path ("files/anvio/ssu/" )dir.create (paste (tmp_path, i, sep = "" ))rm (list = ls (pattern = "tmp_" ))for (i in samp_ps_org) {<- file.path ("files/anvio/ssu/" )dir.create (paste (tmp_path, i, sep = "" ))rm (list = ls (pattern = "tmp_" ))``` ```{r} for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- as.data.frame (t (otu_table (tmp_get)))<- tmp_df %>% rownames_to_column ("Group" )<- file.path ("files/anvio/ssu/" )write.table (tmp_df, paste (tmp_path, i, "/" , "data.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE )rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- transform_sample_counts (tmp_get, function (x) 1e5 * {x/ sum (x)})<- as.data.frame (t (otu_table (tmp_trans)))<- tmp_df %>% rownames_to_column ("Group" )#tmp_name <- purrr::map_chr(i, ~ paste0(., "_trim_tab")) #assign(tmp_name, tmp_df) <- file.path ("files/anvio/ssu/" )write.table (tmp_df, paste (tmp_path, i, "/" , "data_trans.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE )rm (list = ls (pattern = "tmp_" ))``` ### 2. Additional Layers for ASVs ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_indval_final" )))<- tmp_get_indval %>% dplyr:: rename ("Group" = "ASV_ID" ) %>% :: rename ("enrich_indval" = "group" ) %>% :: rename ("test_indval" = "indval" ) %>% :: rename ("pval_indval" = "pval" )<- tmp_get_indval[,1 : 5 ]<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_final" )))<- tmp_get_lefse %>% dplyr:: rename ("Group" = "ASV_ID" ) %>% :: rename ("enrich_lefse" = "group" ) %>% :: rename ("test_lefse" = "lda" ) %>% :: rename ("pval_lefse" = "pval" )<- tmp_get_lefse[,1 : 4 ]<- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- as.data.frame (t (otu_table (tmp_get)))<- cbind (tmp_otu_df, total_reads = rowSums (tmp_otu_df))<- rev (tmp_total)[1 ]<- tmp_total %>% tibble:: rownames_to_column ("Group" )<- as.data.frame (tax_table (tmp_get))$ ASV_SEQ <- NULL $ ASV_ID <- NULL <- tmp_tax_df %>% tibble:: rownames_to_column ("Group" )<- dplyr:: left_join (tmp_tax_df, tmp_total, by = "Group" ) %>% :: left_join (., tmp_get_indval, by = "Group" ) %>% :: left_join (., tmp_get_lefse, by = "Group" )$ ASV_ID <- tmp_add_lay$ Group<- tmp_add_lay[, c (1 ,16 ,8 ,12 ,9 : 11 ,13 : 15 ,2 : 7 )]<- file.path ("files/anvio/ssu/" )write.table (tmp_add_lay, paste (tmp_path, i, "/" , "additional_layers.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE , na = "" )rm (list = ls (pattern = "tmp_" ))``` ### 3. Additional Views for Samples `anvi-import-misc-data` command & `--target-data-table layers` flag.```{r} #| code-fold: true <- read.table ("files/metadata/tables/metadata.txt" ,header = TRUE )<- readRDS ("files/alpha/rdata/ssu18_ps_pime.rds" )data.frame (sample_data (tmp_x))c (2 : 5 )] <- list (NULL )for (i in samp_ps_all) {<- get (i)<- data.frame (sample_data (tmp_get))<- tmp_df[,c (2 : 9 )]<- tmp_df %>% tibble:: rownames_to_column ("id" )<- tmp_df %>% dplyr:: rename ("no_asvs" = "Observed" )<- data.frame (readcount (tmp_get))<- tmp_rc %>% tibble:: rownames_to_column ("id" )<- tmp_rc %>% dplyr:: rename ("no_reads" = 2 )#identical(tmp_df$id, tmp_rc$id) <- dplyr:: left_join (tmp_df, tmp_rc)<- tmp_merge[, c (1 : 6 ,10 ,7 : 9 )]<- dplyr:: left_join (tmp_merge, metadata_tab)<- file.path ("files/anvio/ssu/" )write.table (tmp_final, paste (tmp_path, i, "/" , "additional_views.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE )rm (list = ls (pattern = "tmp_" ))rm (metadata_tab)``` ### 4. Taxon rank abundance by sample [ little nifty block of code ](https://github.com/joey711/phyloseq/issues/418#issuecomment-262637034) written by [ guoyanzhao ](https://github.com/guoyanzhao) on the phyloseq Issues forum, it was a piece of cake. The code can be altered to take any rank. See the post for an explanation.`t_<RANK>!` . For example, `t_class!` should be added for Class rank.`anvi-import-misc-data` command & `--target-data-table layers` flag.```{r} #| code-fold: true <- "Phylum" <- "phylum" for (i in samp_ps_all) {# Make the table <- get (i)<- tax_glom (tmp_get, taxrank = pick_rank)<- psmelt (tmp_glom)<- as.character (tmp_melt[[pick_rank]])<- aggregate (Abundance ~ Sample + tmp_melt[[pick_rank]], tmp_melt, FUN = sum)colnames (tmp_abund)[2 ] <- "tax_rank" library (reshape2)<- as.data.frame (reshape:: cast (tmp_abund, Sample ~ tax_rank))<- tibble:: remove_rownames (tmp_abund)<- tibble:: column_to_rownames (tmp_abund, "Sample" )# Reorder table column by sum <- tmp_abund[,names (sort (colSums (tmp_abund), decreasing = TRUE ))]# Add the prefix <- tmp_layers %>% dplyr:: rename_all (function (x) paste0 ("t_" , pick_rank_l,"!" , x))<- tibble:: rownames_to_column (tmp_layers, "taxon" )# save the table <- file.path ("files/anvio/ssu/" )write.table (tmp_layers, paste (tmp_path, i, "/" , "tax_layers.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE )rm (list = ls (pattern = "tmp_" ))``` ### 5. Construct Dendrograms ```bash anvi-matrix-to-newick data.txt --distance euclidean \ --linkage ward \ -o asv.treanvi-matrix-to-newick data.txt --distance euclidean \ --linkage ward \ -o sample.tre \ --transpose ``` `anvi-matrix-to-newick -h` . Boom.```{r} #| echo: false #bash_commands <- c() USE this to combine all commands # including in loop creates separate files for (i in samp_ps) {<- c ()<- purrr:: map_chr (i, ~ paste0 ("anvi-matrix-to-newick data.txt" ," --distance euclidean --linkage ward -o " ,"asv.tre" ))<- append (bash_commands, tmp_command_asv)<- purrr:: map_chr (i, ~ paste0 ("anvi-matrix-to-newick data.txt" ," --distance braycurtis --linkage complete -o " ,"sample.tre --transpose" ))<- append (bash_commands, tmp_command_samp)<- file.path ("files/anvio/ssu/" )write (bash_commands, paste (tmp_path, i, "/" , "tre.sh" , sep = "" ))rm (list = ls (pattern = "tmp_" ))``` ```{r} #| echo: false # FOR TANSFORMED DATA for (i in samp_ps) {<- c ()<- purrr:: map_chr (i, ~ paste0 ("anvi-matrix-to-newick data_trans.txt" ," --distance euclidean --linkage ward -o " ,"asv_trans.tre" ))<- append (bash_commands, tmp_command_asv)<- purrr:: map_chr (i, ~ paste0 ("anvi-matrix-to-newick data_trans.txt" ," --distance braycurtis --linkage complete -o " ,"sample_trans.tre --transpose" ))<- append (bash_commands, tmp_command_samp)<- file.path ("files/anvio/ssu/" )write (bash_commands, paste (tmp_path, i, "/" , "tre_transformed.sh" , sep = "" ))rm (list = ls (pattern = "tmp_" ))``` ```{bash, echo=FALSE} #NEED to run in anvio from base dir cd ssu18_ps_filtbash tre.shbash tre_transformed.shcd ../cd ssu18_ps_filt_otubash tre.shbash tre_transformed.shcd ../cd ssu18_ps_perfectbash tre.shbash tre_transformed.shcd ../cd ssu18_ps_perfect_otubash tre.shbash tre_transformed.shcd ../cd ssu18_ps_pimebash tre.shbash tre_transformed.shcd ../cd ssu18_ps_pime_otubash tre.shbash tre_transformed.shcd ../``` `phyloseq::distance` and `hclust` . ```{r} #| code-fold: true <- "bray" <- "complete" for (i in samp_ps) {# Make the table <- get (i)<- phyloseq:: distance (physeq = tmp_get, method = pick_dist, type = "sample" )<- hclust (tmp_dist, method = pick_clust)plot (tmp_dend, hang = - 1 )<- as.phylo (tmp_dend) <- file.path ("files/anvio/ssu/" )write.tree (phy = tmp_tree, file = paste (tmp_path, i, "/" , "sample_" , pick_dist, "_" , pick_clust, ".tre" , sep = "" ))rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true <- "euclidean" <- "ward" for (i in samp_ps) {# Make the table <- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- phyloseq:: distance (physeq = tmp_get, method = pick_dist_asv, type = "taxa" )<- hclust (tmp_dist, method = pick_clust_asv)plot (tmp_dend, hang = - 1 )<- as.phylo (tmp_dend) <- file.path ("files/anvio/ssu/" )write.tree (phy = tmp_tree, file = paste (tmp_path, i, "/" , "asv_" , pick_dist_asv, "_" , pick_clust_asv, ".tre" , sep = "" ))rm (list = ls (pattern = "tmp_" ))``` `asv.tre` is **loaded** with `anvi-interactive` command & the `--tree` flag.[ special format ](http://merenlab.org/2017/12/11/additional-data-tables/#layer-orders-additional-data-table) . Specifically, the tree needs to be in a three column, tab delimited, *table*. This way you can add multiple orderings to the same file and view them all in the interactive. The table needs to be in this format:| item_name | data_type | data_value | |-----------|-----------|-----------------------------------------------------| | tree_1 | newick | ((P01_D00_010_W8A:0.0250122,P05_D00_010_W8C:0.02,.. | | tree_2 | newick | ((((((((OTU14195:0.0712585,OTU13230:0.0712585)0:,...| | (…) | (…) | (…) | `sample.tre` is **added** to the profile.db with `anvi-import-misc-data` command & `--target-data-table layer_orders` flag.```{r} #| code-fold: true for (i in samp_ps) {<- file.path ("files/anvio/ssu/" )<- read_file (paste (tmp_path, i, "/" , "sample.tre" , sep = "" ))<- gsub ("[ \r\n ]" , "" , tmp_tree)<- c ("bray_complete" )<- c ("newick" )<- c (tmp_tree)<- data.frame (tmp_item, tmp_type, tmp_df)library (janitor)%>% remove_empty ("rows" )colnames (tmp_tab) <- c ("item_name" , "data_type" , "data_value" )write.table (tmp_tab, paste (tmp_path, i, "/" , "sample.tre" , sep = "" ),sep = " \t " , quote = FALSE , row.names = FALSE , na = "" )rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true # FOR TRANSFORMED DATA for (i in samp_ps) {<- file.path ("files/anvio/ssu/" )<- read_file (paste (tmp_path, i, "/" ,"sample_trans.tre" , sep = "" ))<- gsub ("[ \r\n ]" , "" , tmp_tree)<- c ("bray_complete" )<- c ("newick" )<- c (tmp_tree)<- data.frame (tmp_item, tmp_type, tmp_df)library (janitor)%>% remove_empty ("rows" )colnames (tmp_tab) <- c ("item_name" , "data_type" , "data_value" )write.table (tmp_tab, paste (tmp_path, i, "/" , "sample_trans.tre" , sep = "" ),sep = " \t " , quote = FALSE , row.names = FALSE , na = "" )rm (list = ls (pattern = "tmp_" ))objects ()``` ```{r} #| code-fold: true # FOR HCLUST TREE for (i in samp_ps) {<- file.path ("files/anvio/ssu/" )<- read_file (paste (tmp_path, i, "/" , "sample_" , pick_dist, "_" , pick_clust, ".tre" , sep = "" ))<- gsub ("[ \r\n ]" , "" , tmp_tree)<- c (paste (pick_dist, "_" , pick_clust, "_hclust" , sep = "" ))<- c ("newick" )<- c (tmp_tree)<- data.frame (tmp_item, tmp_type, tmp_df)library (janitor)%>% remove_empty ("rows" )colnames (tmp_tab) <- c ("item_name" , "data_type" , "data_value" )write.table (tmp_tab, paste (tmp_path, i, "/" , "sample_" , pick_dist, "_" , pick_clust, ".tre" , sep = "" ),sep = " \t " , quote = FALSE , row.names = FALSE , na = "" )rm (list = ls (pattern = "tmp_" ))``` ### 6. Make a fasta file `anvi-interactive` command & `--fasta-file` flag.```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- tax_table (tmp_get)<- tmp_tab[, 7 ]<- data.frame (row.names (tmp_tab), tmp_tab)colnames (tmp_df) <- c ("ASV_ID" , "ASV_SEQ" )$ ASV_ID <- sub ("^" , ">" , tmp_df$ ASV_ID)<- file.path ("files/anvio/ssu/" )write.table (tmp_df, paste (tmp_path, i, "/" , i, ".fasta" , sep = "" ),sep = " \n " , col.names = FALSE , row.names = FALSE ,quote = FALSE , fileEncoding = "UTF-8" )rm (list = ls (pattern = "tmp_" ))``` ## Building the Profile Database `--manual` mode, we ***must*** give anvi'o the name of a database and it will *create* that database for us. Then we can shut down the interactive, add the necessary data files, and start back up.```bash anvi-interactive --view-data data.txt \ --tree asv.tre \ --additional-layers additional_layers.txt \ --profile-db profile.db \ --manual ``` `additional_layers.txt` ) and the sample dendrogram (sample.tre) using the command `anvi-import-misc-data` . These commands add the table to the new `profile.db` . First, kill the interactive.```bash anvi-import-misc-data additional_views.txt \ --pan-or-profile-db profile.db \ --target-data-table layersanvi-import-misc-data sample.tre \ --pan-or-profile-db profile.db \ --target-data-table layer_orders``` `tax_layers.txt` ) into the profile database. We will run the same command we just used.```bash anvi-import-misc-data tax_layers.txt \ --pan-or-profile-db profile.db \ --target-data-table layers``` `additional_layers.txt` and import in one command. But we didn't.## Interactive Interface ```bash anvi-interactive --view-data data.txt \ --tree asv.tre \ --additional-layers additional_layers.txt \ --profile-db profile.db--fasta-file anvio.fasta \ --manual ``` ```{r} #| echo: false #| warning: false #| fig-height: 2 :: include_graphics ("figures/16s-da-asvs/before.png" )``` ```{r} #| echo: false # ALL commands #anvi-interactive --view-data data_ssu18_ps_pime.txt --tree asv_ssu18_ps_pime.tre --additional-layers additional_layers_ssu18_ps_pime.txt --profile-db profile_ssu18_ps_pime.db --fasta-file ssu18_ps_pime.fasta --manual #anvi-import-misc-data additional_views_ssu18_ps_pime.txt --pan-or-profile-db profile_ssu18_ps_pime.db --target-data-table layers #anvi-import-misc-data sample_ssu18_ps_pime.tre --pan-or-profile-db profile_ssu18_ps_pime.db --target-data-table layer_orders #anvi-import-misc-data tax_layers_ssu18_ps_pime.txt --pan-or-profile-db profile_ssu18_ps_pime.db --target-data-table layers #anvi-interactive --view-data data_ssu18_ps_pime.txt --tree asv_ssu18_ps_pime.tre --additional-layers additional_layers_ssu18_ps_pime.txt --profile-db profile_ssu18_ps_pime.db --fasta-file ssu18_ps_pime.fasta --manual ``` < details > < summary >< strong > Click here</ strong > for the ITS version of this workflow.</ summary > < br /> ### Visualizing DA ASVs in Anvi’o [ anvi’o ](https://github.com/merenlab/anvio) ---an advanced analysis and visualization platform for ‘omics data [ @eren2015anvi ] ---using the `anvi-interactive` command. Anvi’o likes databases but it also understands that sometimes you do not have a database. So it offers a manual mode. If you type this command you can have a look at the relevant pieces we need for the visualization, specifically those under the headings MANUAL INPUTS and ADDITIONAL STUFF.```bash anvi-interactive -h ``` ``` MANUAL INPUTS: Mandatory input parameters to start the interactive interface without anvi'o databases. --manual-mode We need this flag to run anvi'o in an ad hoc manner, i.e., no database. -f FASTA, --fasta-file FASTA A FASTA-formatted input file. This is sort of optional -d VIEW_DATA, --view-data VIEW_DATA A TAB-delimited file for view data. This is the ASV by sample matrix. We need this -t NEWICK, --tree NEWICK NEWICK formatted tree structure. How the ASVs are ordered in our case. ADDITIONAL STUFF: Parameters to provide additional layers, views, or layer data. -V ADDITIONAL_VIEW, --additional-view ADDITIONAL_VIEW A TAB-delimited file for an additional view to be used in the interface. This file should contain all split names, and values for each of them in all samples. Each column in this file must correspond to a sample name. Content of this file will be called 'user_view', which will be available as a new item in the 'views' combo box in the interface -A ADDITIONAL_LAYERS, --additional-layers ADDITIONAL_LAYERS A TAB-delimited file for additional layer info. In our case this is info about each ASV. The first column of the file must be the ASV names, and the remaining columns should be unique attributes. ``` [ Working with anvi’o additional data tables ](http://merenlab.org/2017/12/11/additional-data-tables/) . A lot of what we need is covered in this tutorial. To get the most out the visualization we need to create a few files to give anvi’o when we fire up the interactive interface.1. View data: in our case, a sample by ASV abundance matrix.2. Additional info about each ASV.3. Additional info about each sample.4. Taxa abundance data for each sample at some rank.5. Dendrograms ordering the ASVs and samples (based on view data).6. Fasta file of all ASVs in the analysis.### Main steps #### 1. View data `-d` or `--view-data` file. This file needs to be an ASV by sample matrix of read counts. To simplify the visualization, we will use ***all*** ASVs represented by 100 or more total reads, including those identified as differentially abundant by the ISA and/or LEfSe. ```{r} #| code-fold: true <- c ("its18_ps_filt" , "its18_ps_pime" , "its18_ps_perfect" , "its18_ps_filt_otu" , "its18_ps_pime_otu" , "its18_ps_perfect_otu" , "its18_ps_work" , "its18_ps_work_otu" )<- c ("its18_ps_filt" , "its18_ps_pime" , "its18_ps_perfect" , "its18_ps_filt_otu" , "its18_ps_pime_otu" , "its18_ps_perfect_otu" )<- c ("its18_ps_work" , "its18_ps_work_otu" )<- c ("its18_ps_pime" , "its18_ps_pime_otu" )<- c ("its18_ps_filt" , "its18_ps_perfect" , "its18_ps_filt_otu" , "its18_ps_perfect_otu" )<- 50 for (i in samp_ps_pime) {<- get (i)<- prune_taxa (taxa_sums (tmp_get) > trim_val, tmp_get)<- purrr:: map_chr (i, ~ paste0 (., "_trim" ))assign (tmp_name, tmp_df)rm (list = ls (pattern = "tmp_" ))<- 300 for (i in samp_ps_other) {<- get (i)<- prune_taxa (taxa_sums (tmp_get) > trim_val, tmp_get)<- purrr:: map_chr (i, ~ paste0 (., "_trim" ))assign (tmp_name, tmp_df)rm (list = ls (pattern = "tmp_" ))objects (pattern = "_trim" )``` ```{r} #| echo: false for (i in samp_ps) {<- file.path ("files/anvio/its/" )dir.create (paste (tmp_path, i, sep = "" ))rm (list = ls (pattern = "tmp_" ))for (i in samp_ps_org) {<- file.path ("files/anvio/its/" )dir.create (paste (tmp_path, i, sep = "" ))rm (list = ls (pattern = "tmp_" ))objects ()``` ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- as.data.frame (t (otu_table (tmp_get)))<- tmp_df %>% rownames_to_column ("Group" )<- file.path ("files/anvio/its/" )write.table (tmp_df, paste (tmp_path, i, "/" , "data.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE )rm (list = ls (pattern = "tmp_" ))objects ()``` ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- transform_sample_counts (tmp_get, function (x) 1e5 * {x/ sum (x)})<- as.data.frame (t (otu_table (tmp_trans)))<- tmp_df %>% rownames_to_column ("Group" )#tmp_name <- purrr::map_chr(i, ~ paste0(., "_trim_tab")) #assign(tmp_name, tmp_df) <- file.path ("files/anvio/its/" )write.table (tmp_df, paste (tmp_path, i, "/" , "data_trans.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE )rm (list = ls (pattern = "tmp_" ))objects ()``` #### 2. Additional Layers for ASVs ```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_indval_final" )))<- tmp_get_indval %>% dplyr:: rename ("Group" = "ASV_ID" ) %>% :: rename ("enrich_indval" = "group" ) %>% :: rename ("test_indval" = "indval" ) %>% :: rename ("pval_indval" = "pval" )<- tmp_get_indval[,1 : 5 ]<- get (purrr:: map_chr (i, ~ paste0 (., "_lefse_final" )))<- tmp_get_lefse %>% dplyr:: rename ("Group" = "ASV_ID" ) %>% :: rename ("enrich_lefse" = "group" ) %>% :: rename ("test_lefse" = "lda" ) %>% :: rename ("pval_lefse" = "pval" )<- tmp_get_lefse[,1 : 4 ]<- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- as.data.frame (t (otu_table (tmp_get)))<- cbind (tmp_otu_df, total_reads = rowSums (tmp_otu_df))<- rev (tmp_total)[1 ]<- tmp_total %>% tibble:: rownames_to_column ("Group" )<- as.data.frame (tax_table (tmp_get))$ ASV_SEQ <- NULL $ ASV_ID <- NULL <- tmp_tax_df %>% tibble:: rownames_to_column ("Group" )<- dplyr:: left_join (tmp_tax_df, tmp_total, by = "Group" ) %>% :: left_join (., tmp_get_indval, by = "Group" ) %>% :: left_join (., tmp_get_lefse, by = "Group" )$ ASV_ID <- tmp_add_lay$ Group<- tmp_add_lay[, c (1 ,16 ,8 ,12 ,9 : 11 ,13 : 15 ,2 : 7 )]<- file.path ("files/anvio/its/" )write.table (tmp_add_lay, paste (tmp_path, i, "/" , "additional_layers.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE , na = "" )rm (list = ls (pattern = "tmp_" ))``` #### 3. Additional Views for Samples `anvi-import-misc-data` command & `--target-data-table layers` flag.```{r} #| code-fold: true <- read.table ("files/metadata/tables/metadata.txt" ,header = TRUE )<- readRDS ("files/alpha/rdata/its18_ps_pime.rds" )data.frame (sample_data (tmp_x))c (2 : 5 )] <- list (NULL )for (i in samp_ps_all) {<- get (i)<- data.frame (sample_data (tmp_get))<- tmp_df[,c (2 : 9 )]<- tmp_df %>% tibble:: rownames_to_column ("id" )<- tmp_df %>% dplyr:: rename ("no_asvs" = "Observed" )<- data.frame (readcount (tmp_get))<- tmp_rc %>% tibble:: rownames_to_column ("id" )<- tmp_rc %>% dplyr:: rename ("no_reads" = 2 )#identical(tmp_df$id, tmp_rc$id) <- dplyr:: left_join (tmp_df, tmp_rc)<- tmp_merge[, c (1 : 6 ,10 ,7 : 9 )]<- dplyr:: left_join (tmp_merge, metadata_tab)<- file.path ("files/anvio/its/" )write.table (tmp_final, paste (tmp_path, i, "/" , "additional_views.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE )rm (list = ls (pattern = "tmp_" ))rm (metadata_tab)``` #### 4. Taxon rank abundance by sample [ little nifty block of code ](https://github.com/joey711/phyloseq/iitses/418#iitsecomment-262637034) written by [ guoyanzhao ](https://github.com/guoyanzhao) on the phyloseq Iitses forum, it was a piece of cake. The code can be altered to take any rank. See the post for an explanation.`t_<RANK>!` . For example, `t_class!` should be added for Class rank.`anvi-import-misc-data` command & `--target-data-table layers` flag.```{r} #| code-fold: true <- "Order" <- "order" for (i in samp_ps_all) {# Make the table <- get (i)<- tax_glom (tmp_get, taxrank = pick_rank)<- psmelt (tmp_glom)<- as.character (tmp_melt[[pick_rank]])<- aggregate (Abundance ~ Sample + tmp_melt[[pick_rank]], tmp_melt, FUN = sum)colnames (tmp_abund)[2 ] <- "tax_rank" library (reshape2)<- as.data.frame (reshape:: cast (tmp_abund, Sample ~ tax_rank))<- tibble:: remove_rownames (tmp_abund)<- tibble:: column_to_rownames (tmp_abund, "Sample" )# Reorder table column by sum <- tmp_abund[,names (sort (colSums (tmp_abund), decreasing = TRUE ))]# Add the prefix <- tmp_layers %>% dplyr:: rename_all (function (x) paste0 ("t_" , pick_rank_l,"!" , x))<- tibble:: rownames_to_column (tmp_layers, "taxon" )# save the table <- file.path ("files/anvio/its/" )write.table (tmp_layers, paste (tmp_path, i, "/" , "tax_layers.txt" , sep = "" ),quote = FALSE , sep = " \t " , row.names = FALSE )rm (list = ls (pattern = "tmp_" ))``` #### 5. Construct Dendrograms ```bash anvi-matrix-to-newick data.txt --distance euclidean \ --linkage ward \ -o asv.treanvi-matrix-to-newick data.txt --distance euclidean \ --linkage ward \ -o sample.tre \ --transpose ``` `anvi-matrix-to-newick -h` . Boom.```{r} #| echo: false #bash_commands <- c() USE this to combine all commands # including in loop creates separate files for (i in samp_ps) {<- c ()<- purrr:: map_chr (i, ~ paste0 ("anvi-matrix-to-newick data.txt" ," --distance euclidean --linkage ward -o " ,"asv.tre" ))<- append (bash_commands, tmp_command_asv)<- purrr:: map_chr (i, ~ paste0 ("anvi-matrix-to-newick data.txt" ," --distance braycurtis --linkage complete -o " ,"sample.tre --transpose" ))<- append (bash_commands, tmp_command_samp)<- file.path ("files/anvio/its/" )write (bash_commands, paste (tmp_path, i, "/" , "tre.sh" , sep = "" ))rm (list = ls (pattern = "tmp_" ))``` ```{r} #| echo: false # FOR TANSFORMED DATA for (i in samp_ps) {<- c ()<- purrr:: map_chr (i, ~ paste0 ("anvi-matrix-to-newick data_trans.txt" ," --distance euclidean --linkage ward -o " ,"asv_trans.tre" ))<- append (bash_commands, tmp_command_asv)<- purrr:: map_chr (i, ~ paste0 ("anvi-matrix-to-newick data_trans.txt" ," --distance braycurtis --linkage complete -o " ,"sample_trans.tre --transpose" ))<- append (bash_commands, tmp_command_samp)<- file.path ("files/anvio/its/" )write (bash_commands, paste (tmp_path, i, "/" , "tre_transformed.sh" , sep = "" ))rm (list = ls (pattern = "tmp_" ))``` ```{bash, echo=FALSE} #NEED to run in anvio from base dir cd its18_ps_filtbash tre.shbash tre_transformed.shcd ../cd its18_ps_filt_otubash tre.shbash tre_transformed.shcd ../cd its18_ps_perfectbash tre.shbash tre_transformed.shcd ../cd its18_ps_perfect_otubash tre.shbash tre_transformed.shcd ../cd its18_ps_pimebash tre.shbash tre_transformed.shcd ../cd its18_ps_pime_otubash tre.shbash tre_transformed.shcd ../``` `phyloseq::distance` and `hclust` . ```{r} #| code-fold: true <- "bray" <- "complete" for (i in samp_ps) {# Make the table <- get (i)<- phyloseq:: distance (physeq = tmp_get, method = pick_dist, type = "sample" )<- hclust (tmp_dist, method = pick_clust)plot (tmp_dend, hang = - 1 )<- as.phylo (tmp_dend) <- file.path ("files/anvio/its/" )write.tree (phy = tmp_tree, file = paste (tmp_path, i, "/" , "sample_" , pick_dist, "_" , pick_clust, ".tre" , sep = "" ))rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true <- "euclidean" <- "ward" for (i in samp_ps) {# Make the table <- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- phyloseq:: distance (physeq = tmp_get, method = pick_dist_asv, type = "taxa" )<- hclust (tmp_dist, method = pick_clust_asv)plot (tmp_dend, hang = - 1 )<- as.phylo (tmp_dend) <- file.path ("files/anvio/its/" )write.tree (phy = tmp_tree, file = paste (tmp_path, i, "/" , "asv_" , pick_dist_asv, "_" , pick_clust_asv, ".tre" , sep = "" ))rm (list = ls (pattern = "tmp_" ))``` `asv.tre` is **loaded** with `anvi-interactive` command & the `--tree` flag.[ special format ](http://merenlab.org/2017/12/11/additional-data-tables/#layer-orders-additional-data-table) . Specifically, the tree needs to be in a three column, tab delimited, *table*. This way you can add multiple orderings to the same file and view them all in the interactive. The table needs to be in this format:| item_name | data_type | data_value | |-----------|-----------|-----------------------------------------------------| | tree_1 | newick | ((P01_D00_010_W8A:0.0250122,P05_D00_010_W8C:0.02,.. | | tree_2 | newick | ((((((((OTU14195:0.0712585,OTU13230:0.0712585)0:,...| | (…) | (…) | (…) | `sample.tre` is **added** to the profile.db with `anvi-import-misc-data` command & `--target-data-table layer_orders` flag.```{r} #| code-fold: true for (i in samp_ps) {<- file.path ("files/anvio/its/" )<- read_file (paste (tmp_path, i, "/" , "sample.tre" , sep = "" ))<- gsub ("[ \r\n ]" , "" , tmp_tree)<- c ("bray_complete" )<- c ("newick" )<- c (tmp_tree)<- data.frame (tmp_item, tmp_type, tmp_df)library (janitor)%>% remove_empty ("rows" )colnames (tmp_tab) <- c ("item_name" , "data_type" , "data_value" )write.table (tmp_tab, paste (tmp_path, i, "/" , "sample.tre" , sep = "" ),sep = " \t " , quote = FALSE , row.names = FALSE , na = "" )rm (list = ls (pattern = "tmp_" ))``` ```{r} #| code-fold: true # FOR TRANSFORMED DATA for (i in samp_ps) {<- file.path ("files/anvio/its/" )<- read_file (paste (tmp_path, i, "/" ,"sample_trans.tre" , sep = "" ))<- gsub ("[ \r\n ]" , "" , tmp_tree)<- c ("bray_complete" )<- c ("newick" )<- c (tmp_tree)<- data.frame (tmp_item, tmp_type, tmp_df)library (janitor)%>% remove_empty ("rows" )colnames (tmp_tab) <- c ("item_name" , "data_type" , "data_value" )write.table (tmp_tab, paste (tmp_path, i, "/" , "sample_trans.tre" , sep = "" ),sep = " \t " , quote = FALSE , row.names = FALSE , na = "" )rm (list = ls (pattern = "tmp_" ))objects ()``` ```{r} #| code-fold: true # FOR HCLUST TREE for (i in samp_ps) {<- file.path ("files/anvio/its/" )<- read_file (paste (tmp_path, i, "/" , "sample_" , pick_dist, "_" , pick_clust, ".tre" , sep = "" ))<- gsub ("[ \r\n ]" , "" , tmp_tree)<- c (paste (pick_dist, "_" , pick_clust, "_hclust" , sep = "" ))<- c ("newick" )<- c (tmp_tree)<- data.frame (tmp_item, tmp_type, tmp_df)library (janitor)%>% remove_empty ("rows" )colnames (tmp_tab) <- c ("item_name" , "data_type" , "data_value" )write.table (tmp_tab, paste (tmp_path, i, "/" , "sample_" , pick_dist, "_" , pick_clust, ".tre" , sep = "" ),sep = " \t " , quote = FALSE , row.names = FALSE , na = "" )rm (list = ls (pattern = "tmp_" ))``` #### 6. Make a fasta file `anvi-interactive` command & `--fasta-file` flag.```{r} #| code-fold: true for (i in samp_ps) {<- get (purrr:: map_chr (i, ~ paste0 (., "_trim" )))<- tax_table (tmp_get)<- tmp_tab[, 7 ]<- data.frame (row.names (tmp_tab), tmp_tab)colnames (tmp_df) <- c ("ASV_ID" , "ASV_SEQ" )$ ASV_ID <- sub ("^" , ">" , tmp_df$ ASV_ID)<- file.path ("files/anvio/its/" )write.table (tmp_df, paste (tmp_path, i, "/" , i, ".fasta" , sep = "" ),sep = " \n " , col.names = FALSE , row.names = FALSE ,quote = FALSE , fileEncoding = "UTF-8" )rm (list = ls (pattern = "tmp_" ))``` ### Building the Profile Database `--manual` mode, we ***must*** give anvi'o the name of a database and it will *create* that database for us. Then we can shut down the interactive, add the necessary data files, and start back up.```bash anvi-interactive --view-data data.txt \ --tree asv.tre \ --additional-layers additional_layers.txt \ --profile-db profile.db \ --manual ``` `additional_layers.txt` ) and the sample dendrogram (sample.tre) using the command `anvi-import-misc-data` . These commands add the table to the new `profile.db` . First, kill the interactive.```bash anvi-import-misc-data additional_views.txt \ --pan-or-profile-db profile.db \ --target-data-table layersanvi-import-misc-data sample.tre \ --pan-or-profile-db profile.db \ --target-data-table layer_orders``` `tax_layers.txt` ) into the profile database. We will run the same command we just used.```bash anvi-import-misc-data tax_layers.txt \ --pan-or-profile-db profile.db \ --target-data-table layers``` `additional_layers.txt` and import in one command. But we didn't.### Interactive Interface ```bash anvi-interactive --view-data data.txt \ --tree asv.tre \ --additional-layers additional_layers.txt \ --profile-db profile.db--fasta-file anvio.fasta \ --manual ``` ```{r} #| echo: false #| warning: false #| fig-height: 2 :: include_graphics ("figures/16s-da-asvs/before.png" )``` ```{r} #| echo: false # ALL commands #anvi-interactive --view-data data_its18_ps_pime.txt --tree asv_its18_ps_pime.tre --additional-layers additional_layers_its18_ps_pime.txt --profile-db profile_its18_ps_pime.db --fasta-file its18_ps_pime.fasta --manual #anvi-import-misc-data additional_views_its18_ps_pime.txt --pan-or-profile-db profile_its18_ps_pime.db --target-data-table layers #anvi-import-misc-data sample_its18_ps_pime.tre --pan-or-profile-db profile_its18_ps_pime.db --target-data-table layer_orders #anvi-import-misc-data tax_layers_its18_ps_pime.txt --pan-or-profile-db profile_its18_ps_pime.db --target-data-table layers #anvi-interactive --view-data data_its18_ps_pime.txt --tree asv_its18_ps_pime.tre --additional-layers additional_layers_its18_ps_pime.txt --profile-db profile_its18_ps_pime.db --fasta-file its18_ps_pime.fasta --manual ``` </ details > ```{r} #| echo: false save.image ("page_build/mmarker_wf.rdata" )``` ```{r} #| include: false #| eval: true remove (list = ls ())``` # Workflow Output < iframe src = "https://widgets.figshare.com/articles/16828249/embed?show_title=1" width = "100%" height = "251" allowfullscreen frameborder = "0" ></ iframe > </ br > < a href = "metadata.html" class = "btnnav button_round" style = "float: right;" > Next workflow:< br > 8. Metadata Analysis</ a > < a href = "beta.html" class = "btnnav button_round" style = "float: left;" > Previous workflow:< br > 6. Beta Diversity & Dispersion Estimates</ a >< br >< br > ```{r} #| message: false #| results: hide #| eval: true #| echo: false remove (list = ls ())### COmmon formatting scripts ### NOTE : captioner.R must be read BEFORE captions_XYZ.R source (file.path ("assets" , "functions.R" ))``` #### Source Code {.appendix} `r fa(name = "github")` by [ clicking this link ] (`r source_code()` ). #### Data Availability {.appendix} [ 10.25573/data.16828249 ](https://doi.org/10.25573/data.16828249) .#### Last updated on {.appendix} ```{r} #| echo: false #| eval: true Sys.time ()```